
Abstract
Background We sought to investigate how penetrance of familial cancer syndromes varies with family history using a population-based cohort.
Methods We analysed 454,712 UK Biobank participants with exome sequence and clinical data. We identified participants with a self-reported family history of breast or colorectal cancer and a pathogenic/likely pathogenic variant in the major genes responsible for hereditary breast cancer or Lynch syndrome. We calculated survival to cancer diagnosis (controlled for age, sex, death, recruitment centre, screening and prophylactic surgery).
Results Women with a pathogenic BRCA1 or BRCA2 variant had an increased risk of breast cancer that was significantly higher in those with a first-degree family history (relative hazard 10.29 and 7.82, respectively) than those without (7.82 and 4.66). Penetrance to age 60 was also higher in those with a family history (44.7% and 24.1%) versus those without (22.8% and 17.9%). A similar pattern was seen in Lynch syndrome: individuals with a pathogenic MLH1, MSH2 or MSH6 variant had an increased risk of bowel cancer that was significantly higher in those with a family history (relative hazard 63.7, 68.4 and 12.1) than those without (20.9, 18.6 and 5.9). Penetrance to age 60 was also higher for carriers of a pathogenic MLH1 or MSH2 variant in those with a family history (27.1% and 25.2%) versus those without (15.2% and 3.2%).
Conclusions Individuals with pathogenic cancer syndrome variants are at significantly less elevated risk of cancer in the absence of family history (risk ratio 0.57), so invasive follow-up may be unwarranted.
Introduction
Genetic testing for inherited cancer syndromes is offered to affected individuals based on various qualifying criteria1,2. For example, BRCA1 and BRCA2 testing is offered to individuals with breast or ovarian cancer and/or a known history of hereditary breast and ovarian cancer (HBOC)3 in many countries, including the UK and USA, although there have been suggestions that this should be expanded to all women diagnosed with breast cancer4. Similarly, patients presenting with bowel cancer are routinely offered screening for hereditary non-polyposis colorectal cancer (HNPCC, or Lynch syndrome)5. If a causal pathogenic variant is identified, chemoprevention or prophylactic mastectomy/oophorectomy may be advised for HBOC, or regular colonoscopies or prophylactic surgery for Lynch syndrome6,7. However, if no causal variant is found, individuals with a family history of breast or bowel cancer still have a higher cancer risk relative to the wider population, indicating as yet unknown variants, genes or further risk factors are involved8,9.
Traditionally, clinical genetics has used a phenotype-first approach to identify the most likely cases with an underlying genetic pathology10. Some pathogenic variants in genetic conditions have incomplete penetrance (where only a proportion of variant carriers will develop the condition), leading to reduced risk of disease11. In the context of hereditary cancer syndromes, by offering genetic testing only to those individuals who have a family history of cancer, there is an inherent ascertainment bias towards finding highly penetrant variants in those families; the variant must have segregated through the family and been detected in an individual with cancer. This bias can lead to artificially high estimates of the penetrance of some variants. However, if found incidentally in an individual with no family history of disease, the penetrance of the same pathogenic variant is unlikely to be as high12.
Increasingly, a genotype-first approach is being used to identify individuals with pathogenic variants13-15. The challenges of reporting and interpreting variants discovered in the absence of a phenotype have been explored16. Population sampling of unselected individuals is required for calculating prevalence and penetrance of genetic variants17, and population databases have already been central to confirming or refuting pathogenicity of genetic variants and validating clinical decisions18. The extent to which family-based penetrance impacts variant pathogenicity through unknown risk modifiers remains unquantified, though these modifying effects are being studied in cancer cohorts19. Uncertainty surrounding penetrance estimates between familial disease cohorts and unselected population cohorts could lead to the provision of inaccurate information and unwarranted interventions which are not evidence-based20.
Here we use exome sequencing data and clinical records from 454,712 individuals from UK Biobank to estimate the population penetrance of pathogenic genetic variants for two cancer-predisposition syndromes (HBOC and Lynch syndrome) and investigate the effect of family history of disease on these estimates.
Methods
Cohort
We used data from UK Biobank21. Hospital Episode Statistics and cancer registry data were available for the whole cohort up to 25 June 2021, and baseline participant questionnaires. Exome sequencing data were available on 454,712 individuals (219,134 women), generated externally by Regeneron22. The UK Biobank resource was approved by the UK Biobank Research Ethics Committee and all participants provided written informed consent to participate.
Variant identification
Detailed sequencing methodology for UK Biobank samples is provided by Szustakowski et al.23, exomes were captured with the IDT xGen Exome Research Panel v1.0 which targeted 39Mbp of the human genome with coverage exceeds on average 20x on 95.6% of sites. The OQFE protocol was used for mapping and variant calling to the GRCh38 reference. We included variants that had individual and variant missingness <10%, Hardy Weinberg Equilibrium p-value >10−15, minimum read depth of 7 for SNVs and 10 for indels, and at least one sample per site passed the allele balance threshold > 15% for SNVs and 20% for indels. Variants were annotated using the Ensembl Variant Effect Predictor (VEP)24.
Pathogenic variant classification
Variants were considered in clinically relevant transcripts for HBOC in the BRCA1 (ENST00000357654) and BRCA2 (ENST00000380152) genes (hereafter collectively referred to as BRCA variants) and for hereditary bowel cancer in the MLH1 (ENST00000231790), MSH2 (ENST00000233146) and MSH6 (ENST00000234420) genes. As described previously25, variants in these genes were defined as pathogenic if they had been classified as pathogenic or likely pathogenic at 2* level or above in the ClinVar database (accessed April 2022)26. We also included likely protein truncating variants (PTV), which we defined as any variant that is predicted to cause a premature stop gain, a frameshift, or abolish a canonical splice site (−2 or +2 bp from exon boundary); we excluded PTVs in the last exon of each gene. Any pathogenic variants identified were confirmed visually using the Integrative Genomics Viewer (IGV)27 to examine the alignments of quality controlled sequencing reads, and likely false positive variants were excluded.
Cancer diagnosis and age at diagnosis
Cancer registry data for breast and bowel cancer was collected for all UK Biobank participants with exome sequencing data. Although both BRCA and Lynch syndrome variants are linked to multiple other cancers, family history information was only available for breast and bowel cancer and so the analysis was limited to these cancer types. ICD-9 and ICD-10 codes were used to identify individuals with breast cancer (ICD-9 codes 174 (all subcodes); ICD-10 codes C50 (all subcodes)) and bowel cancer (ICD-9 codes 153 (all subcodes; ICD-10 codes C18 (all subcodes). The age at diagnosis was also extracted from the registry for these individuals.
Family history calculation
UK Biobank participants were asked about 12 specific illnesses within their family as part of the enrolment process. We used the fields 20107 (illnesses of father), 20110 (illnesses of mother) and 20111 (illnesses of siblings) to create a new variable of positive first-degree family history for breast and bowel cancer.
Statistical testing
All data analysis and statistical testing was performed in Stata. Survival analysis with Cox logistic regressions were carried out to assess the relationship between individuals with a pathogenic BRCA or Lynch syndrome variant and first-degree family history. BRCA analysis was restricted to women. A Cox linear regression model was built using mastectomy in the absence of cancer (to control for prophylactic treatment biasing outcome in BRCA carriers), age, breast or bowel screening, death, recruitment centre and sex (Lynch analysis only). Sub-group analyses were also performed on those groups stratified by positive or negative first-degree family history. The resultant model was used to predict survival functions.
Kaplan-Meier curves were generated from the age when individuals were diagnosed with breast or bowel cancer. Cox proportional hazard tests for equivalence of survival functions were used to interrogate inter-group differences. Incidence-rate ratios were also calculated.
Results
Women with a pathogenic BRCA variant and a family history of breast cancer have a significantly increased risk of breast cancer compared to those with a pathogenic BRCA variant alone
We identified 230 women with a pathogenic variant in BRCA1 (BRCA1+) and 611 in BRCA2 (BRCA2+). Carriers were further categorised into those who had a first-degree relative with breast cancer (FH+; n=78 for BRCA1 and n=170 for BRCA2) and those who did not (FH-; n=152 for BRCA1 and n=441 for BRCA2).
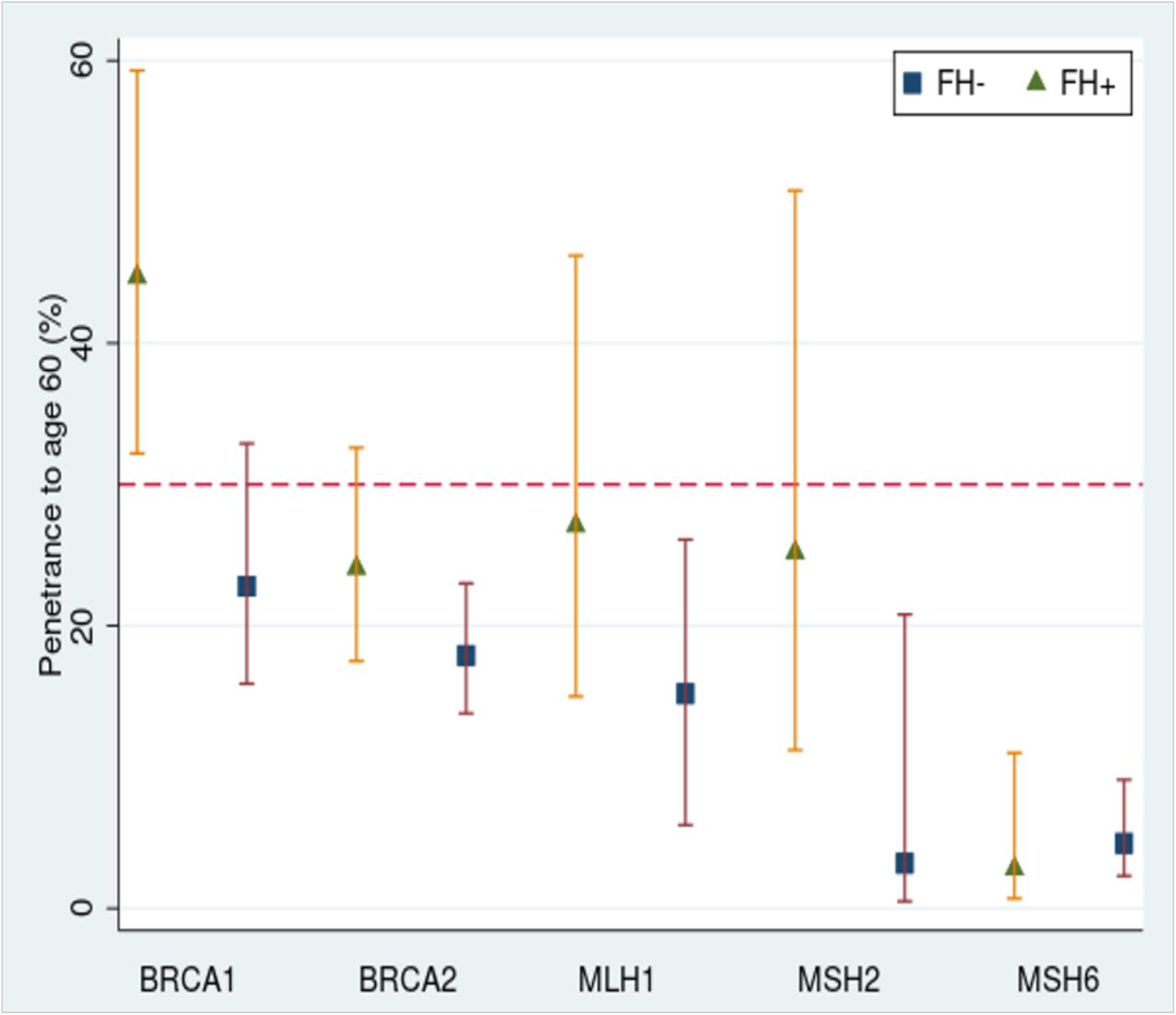
Kaplan-Meier curves were generated for the different groups (i.e. BRCA +/- and family history +/-) and the Cox regression-based test for equality of survival curves demonstrated a significant difference between variant carriers with and without family history for both BRCA1 (chi2 = 604.27, p<0.0001, relative hazard 10.29 with family history and 7.24 without) and BRCA2 (chi2 = 689.63, p<0.0001, relative hazard 7.82 with family history and 4.66 without) (Figure 1). The survival model predicts a significantly increased penetrance (chi2 =11.7 p<0.001) to age 60 in BRCA1+/FH+ women (44.7% 95% CI 32.2-59.3) compared to BRCA1+/FH-women (22.8% 95% CI 15.9-32.0). The predicted penetrance to age 60 in BRCA2+/FH+ women was 24.1% (95% CI 17.5-32.6) versus 17.9% (95% CI 13.8-23.0) in BRCA2+/FH-women. Incidence-rates were also significantly higher in both BRCA1+/FH+ (rate ratio 1.5 p=0.04) and BRCA2+/FH+ (rate ratio 1.7 p<0.0001) women compared to those who were FH-(Figure 2).

Participants were stratified and Kaplan-Meier survival curves plotted based on whether they had a pathogenic BRCA1 (women only) (A), BRCA2 (B) (women only), MLH1 (C), MSH2 (D) or MSH6 (E) variant and/or first-degree family history of breast cancer.

Penetrance to age 60 was calculated using the survival model and shown per gene, split by positive or negative family history. A red dotted line has been added at 30% to indicate the lifetime risk level (note: not risk to age 60) used by NICE to guide enhanced surveillance in women at risk of breast cancer in the UK29. These guidelines consider all BRCA1 and BRCA2 carriers to be high risk. The latest Mallorca guidelines for Lynch syndrome also suggest enhanced surveillance for all MLH1, MSH2 and MSH6 carriers37.
Individuals with a pathogenic Lynch variant and a family history of bowel cancer have an increased risk of bowel cancer compared to those with a pathogenic Lynch variant alone
We identified 89 individuals with a pathogenic variant in MLH1 (MLH1+), 71 in MSH2 (MSH2+) and 421 in MSH6 (MSH6+). Carriers were further categorised into those who had a first-degree relative with bowel cancer (FH+; n=45 for MLH1, n=39 for MHS2 and n=114 for MSH6) and those who did not (FH-; n=44 for MLH1, n=32 for MHS2 and n=307 for MSH6).
Kaplan-Meier curves were generated for the different groups and the Cox regression-based test for equality of survival curves demonstrated a significant difference between variant carriers with and without family history for MLH1 (chi2 = 205.3, p<0.0001, relative hazard 63.7 with a family history and 20.9 without), MSH2 (chi2 = 153.28, p<0.0001, relative hazard 68.4 with a family history and 18.6 without) and MSH6 (chi2 = 130.1 p<0.0001, relative hazard 12.1 with a family history and 5.9 without) (Figure 1). The survival model predicts an increased penetrance to age 60 in MLH1+/FH+ individuals (27.1% 95% CI 15.0-46.2) compared to MLH1+/FH-individuals (15.2% 95% CI 5.9-26.1). For MSH2+ individuals, the penetrance to age 60 was 25.2% (95% CI 11.2-50.9) for FH+ and 3.2% (95% CI 0.5-20.8) for FH-; for MSH6+ individuals, the penetrance to age 60 was 2.8% (95% CI 0.7-11.0) for FH+ and 4.6% (95% CI 2.3-9.1) for FH- (Figure 2). Incidence-rate comparison showed a significant difference for MLH1+/FH+ (rate ratio 2.4 p=0.03) and MSH2+/FH+ (rate ratio 2.2 p=0.01) but not MSH6+/FH+ individuals (rate ratio 2.0 p=0.05) compared to FH-.
Combined analysis across all genes showed a consistent trend for significantly elevated risk of cancer in pathogenic variant carriers with a family history versus those without
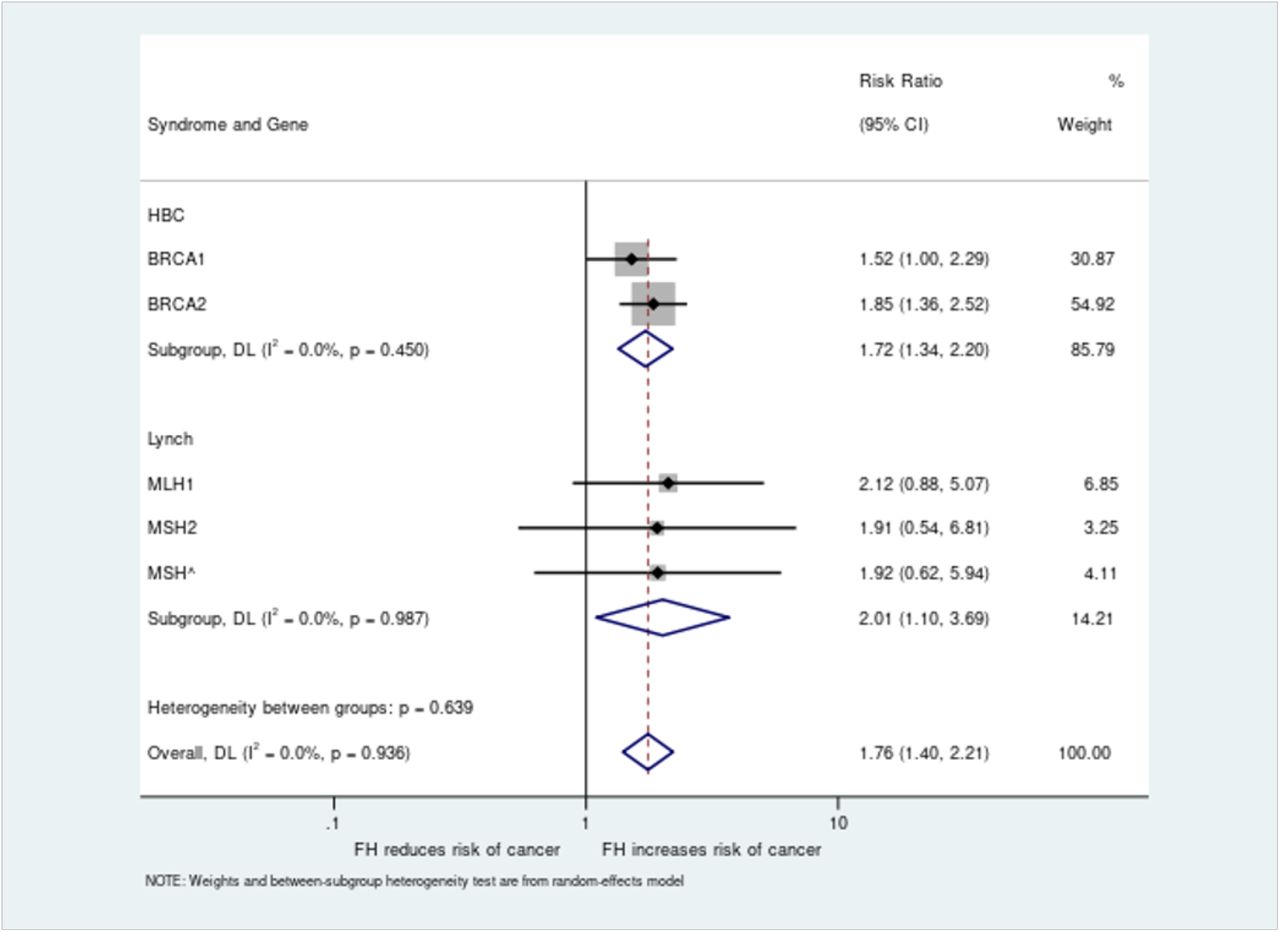
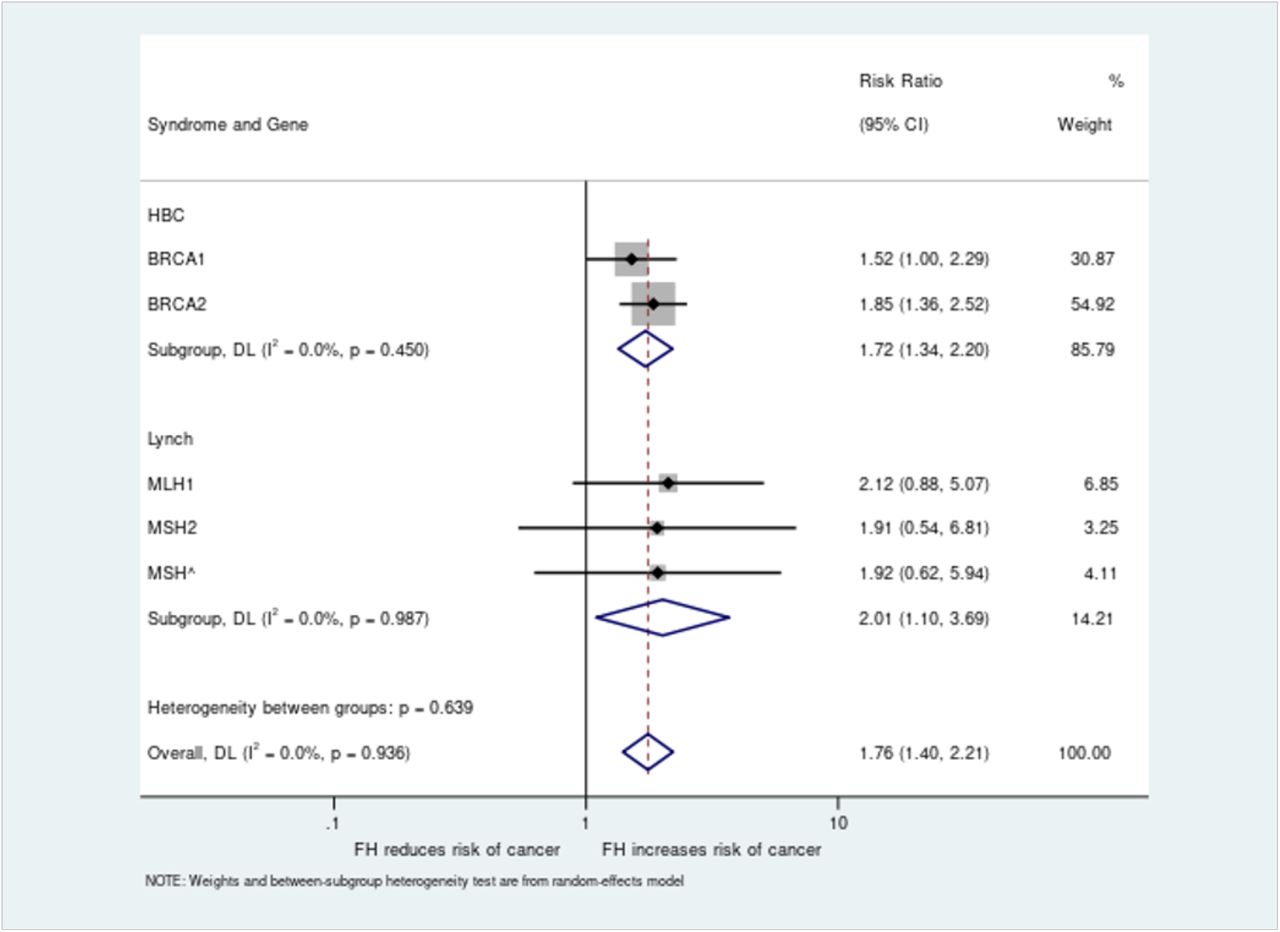
Relative risk values were generated for all five genes and combined analysis performed across both syndromes for all pathogenic variant carriers, giving an overall increased risk of cancer in those with a family history of 1.76 (95% CI 1.40-2.21) versus those without (Figure 3). Subgroup analyses for HBOC (BRCA1/BRCA2) and Lynch (MLH1/MSH2/MSH6) were also significant, with relative risks of family history of 1.74 (95% CI 1.34-2.20) and 2.01 (95% CI 1.10-3.69) respectively.
{kind=link}
{kind=link}
{kind=link}
The risk ratio for cancer diagnosis based on presence or absence of family history was analysed for women with BRCA1 or BRCA2 pathogenic variants and all individuals with MLH1, MSH2 or MSH6 variants. Individual gene level analysis was performed along with combined analyses per syndrome and across all genes.
Discussion
Risk of breast or bowel cancer in pathogenic variant carriers is significantly less elevated in the absence of family history
Using data from a large population cohort, we have shown that much of the risk conferred by a rare pathogenic variant associated with HBOC or Lynch syndrome is conferred by a first-degree family history of disease. In UK Biobank, women with a BRCA1/2 variant are 1.5/1.9-times more likely to get breast cancer if they also have a first-degree family history of breast cancer, whilst individuals with a MLH1/MSH2/MSH6 variant are 2.1/1.9/1.9-times more likely to get bowel cancer if they also have a first-degree family history of bowel cancer. Carriers with a family history are also more likely to develop cancer earlier versus those without. These risk increases are consistent with those previously observed in clinically ascertained cohorts28 but have not previously been estimated in a population cohort. This difference in penetrance in carrier individuals could be sufficient to justify stratifying just individuals with a family history into high-risk groups currently eligible for specialist clinical care (e.g. NICE guidance on familial breast cancer requires >30% lifetime risk to be eligible for referral to a specialist genetic clinic in the UK)29.
Our results are consistent with comparable population studies of cancer susceptibility, but to the best of our knowledge, our study is the first to investigate the effect of family history alone in a clinically unselected population across multiple syndromes. The penetrance of BRCA1/2 mutations was previously estimated in 49,960 individuals in UK Biobank, but the analysis did not evaluate family history30. Among pathogenic variant carriers for both HBOC and Lynch syndrome, the probability of disease by age 75 has been estimated to range from 13-76% for breast cancer and 11-80% for colon cancer respectively, based on different polygenic background, but again this analysis did not specifically analyse the effect of having a first-degree relative with the disease31. Recent work in a smaller subset of UK Biobank has also shown consistent results in colorectal cancer, highlighting the added value of family history in combination with polygenic risk scores32. The penetrance of HBOC amongst clinically unselected pathogenic BRCA1/2 variant carriers was previously shown to be significantly different between those with and without a family history (83% versus 60% to age 60 for BRCA1, and 76% versus 33% to age 80 for BRCA2), but the study was limited to just three variants that are relatively common in the Ashkenazi Jewish population of Israel33.
Ascertainment bias and other limitations may impact the generalisability of findings made through population biobanks
Whilst existing family-based cohorts suffer from an ascertainment bias that is likely to overinflate penetrance estimates, the older cohort in the UK Biobank is likely to be confounded by survival bias34, i.e. individuals with the most severe early-onset disease will not appear as they would have died prior to recruitment. Despite taking a conservative approach to pathogenic variant classification, this bias will have the effect of removing very highly penetrant variants from the cohort, which is likely to deflate penetrance estimates. The true penetrance for an unselected population is likely to be somewhere between the figures generated from each context. The UK Biobank is also not a representative population cohort, due to recognised recruitment biases35, and so these estimates are likely to represent a lower bound.
Additionally, a full family history was not recorded for participants in UK Biobank, so we relied upon self-reported illness in first-degree relatives as a proxy for family history and were only able to include breast and bowel cancers. Given the age of the UK Biobank cohort (recruited from 40-69 years old), these recollections could be biased towards more aggressive or early-onset disease, particularly in parents.
Finally, due to the rarity of individual pathogenic variants, we were limited by the size of the existing cohort. We have therefore included data from all eligible participants in UK Biobank (n=454,712), regardless of ethnicity or consanguinity. We are underpowered to detect differences in penetrance between individual variants, which will also be important for risk discussions with patients. Future research with larger cohorts is needed further improve risk prediction and investigate modifiers.
Implications for practice
The findings of this study suggest that any universal policy of returning pathogenic cancer-predisposing genetic variants found incidentally or through direct-to-consumer genetic testing of asymptomatic individuals should be approached with caution. It will be very difficult to counsel individuals as to their particular risk profile without further pedigree construction or investigations. If penetrance estimates from affected families are used, there is a danger of over-management of asymptomatic individuals with no family history of disease. These “patients-in-waiting” may be exposed to unnecessary surveillance or more invasive prophylactic procedures6,7. Follow-up data gathered from such initiatives as the UK 100,000 Genomes Project13 and the American College of Medical Genetics and Genomics Recommendations on Reporting of Secondary Findings14 will be critical to decipher the exact risk profile of unselected variant carriers. It is imperative that individuals who receive genotype information indicating a predisposition to cancer are appropriately counselled as to their individual risk profile in the context of their family history of disease. For those ascertained outside of the standard clinical pathway, this will help to avoid patients at risk levels not far above background making injudicious decisions about prophylactic options.
Conclusion
It has long been known, though is not widely appreciated, that penetrance estimates for pathogenic variants causing hereditary subtypes of common diseases are likely to be significantly inflated due to ascertainment bias12. The use of selection criteria for genetic testing based on multiple affected family members3,36 will necessarily bias the findings towards those families in whom the variants have a high penetrance. We have shown that, even in a clinically unselected population, having an affected first-degree relative significantly increases the penetrance of pathogenic variants in two hereditary cancer syndromes. Systematic testing either of all patients with breast or bowel cancer or in truly unselected populations is likely to yield more conservative estimates.
Data Availability
All data produced in the present study are available upon reasonable request to the authors
Funding
The current work is supported by the MRC (grant no MR/T00200X/1). The MRC had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Declaration of interests
The authors declare no competing interests.
Acknowledgements
This research has been conducted using the UK Biobank Resource under Application Number 49847. The authors would like to acknowledge the use of the University of Exeter High-Performance Computing (HPC) facility in carrying out this work.
References



