
Abstract
New and more transmissible variants of SARS-CoV-2 have arisen multiple times over the course of the pandemic. Rapidly identifying mutations that affect transmission could facilitate outbreak control efforts and highlight new variants that warrant further study. Here we develop an analytical epidemiological model that infers the transmission effects of mutations from genomic surveillance data. Applying our model to SARS-CoV-2 data across many regions, we find multiple mutations that strongly affect the transmission rate, both within and outside the Spike protein. We also quantify the effects of travel and competition between different lineages on the inferred transmission effects of mutations. Importantly, our model detects lineages with increased transmission as they arise. We infer significant transmission advantages for the Alpha and Delta variants within a week of their appearances in regional data, when their regional frequencies were only around 1%. Our model thus enables the rapid identification of variants and mutations that affect transmission from genomic surveillance data.
Viruses can acquire mutations that affect how efficiently they infect new hosts, for example by increasing viral load or escaping host immunity1–4. The ability to rapidly identify mutations that increase transmission could inform outbreak control efforts and identify potential immune escape variants5–9. However, estimating how individual mutations affect viral transmission is a challenging problem.
Current methods to estimate changes in viral transmission generally rely on phylogenetic analyses or fitting changes in variant frequencies to a simple growth model5,10–12. Phylogenetic analyses for viruses can be challenging due to a high degree of sequence similarity, which implies that the data can be explained equally well by a number of different trees13. Phylogenetic analyses also typically rely on extensive Markov chain Monte Carlo sampling that becomes intractable for very large data sets. Simple growth models can estimate the difference in transmissibility between one variant and others circulating in the same region. However, they typically do not systematically account for competition between multiple variants, and their estimates may be difficult to compare for variants that arose in other regions or with different genetic backgrounds. These approaches also do not consider travel of infected individuals, nor do they account for superspreading —where a small number of infected individuals cause the majority of secondary infections —which has been observed for viruses like SARS-CoV and SARS-CoV-214,15.
To overcome these challenges, we developed a method to infer the effects of single nucleotide variants (SNVs) on viral transmission from genomic surveillance data that accounts for competition between viral lineages, travel, superspreading, and outbreaks in different locations. For clarity, we refer to non-reference nucleotides (including deletions or insertions) as SNVs and viral lineages possessing common sets of SNVs as variants. Simulations show that our approach can reliably estimate transmission effects of SNVs even from limited data. We applied our method to more than 1.6 million SARS-CoV-2 sequences from 87 geographical regions to reveal the effects of mutations on viral transmission throughout the pandemic. While the vast majority of SARS-CoV-2 mutations have negligible effects, we readily observe increased transmission for sets of SNVs in Spike and other hotspots throughout the genome. We further quantified the influence of travel and competition between multiple variants, using Nextstrain lineage 20E (EU1) as an example case. We found that realistic rates of travel during the pandemic would only slightly affect estimated changes in viral transmission. However, competition between variants has significant effects that must be accounted for in order to accurately estimate changes in transmission.
Importantly, our approach is sensitive enough to identify variants with increased transmission before they reach high frequencies. We demonstrate our capacity for early detection by studying the rise of the Alpha and Delta variants using data from the UK. In both cases, we reliably infer increased transmission for these variants within a week of their emergence, when their frequency in the region was only around 1%. Collectively, these data show that our model can be applied for the surveillance of evolving pathogens to robustly identify variants with transmission advantages and to highlight key mutations that may be driving changes in transmission.
Results
Epidemiological Model
To quantify the effects of mutations on viral transmission, we developed a stochastic branching process model of infection based on the well-known susceptible-infected-recovered (SIR) model. Our model incorporates superspreading by drawing the number of secondary infections caused by an infected individual from a negative binomial distribution with mean R, referred to as the effective reproduction number, and dispersion parameter k (refs.14,15). Multiple variants with different transmission rates are included by assigning a variant a an effective reproduction number Ra = R(1 +wa). Under an additive model, the net increase or decrease in transmission for a variant is the sum of the individual transmission effects si for each SNV i that the variant contains. In analogy with population genetics, we refer to the wa and si as selection coefficients. In addition to superspreading and multiple variants, our model also incorporates travel of infected individuals into or out of a localized outbreak (Methods).
We can then apply Bayesian inference to estimate the transmission effects of SNVs that best explain the observed evolutionary history of an outbreak. To simplify our analysis, we use a path integral technique from statistical physics, recently applied in the context of population genetics16, to efficiently quantify the probability of the model parameters given the data (for details, see Supplementary Information). This allows us to derive an analytical estimate for the maximum a posteriori selection coefficients ŝ for a given set of viral genomic surveillance data,
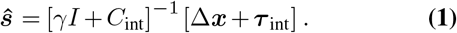 Here Δx is the change in the SNV frequency vector over time, γ specifies the width of a Gaussian prior probability distribution for the selection coefficients si, and I is the identity matrix. Cint is the covariance matrix of SNV frequencies integrated over time, and accounts for competition between variants as well as the speed of growth for different viral lineages (Supplementary Information). τint accounts for travel and is given explicitly in equation (S2). Data from multiple outbreaks can be combined by summing contributions to the integrated covariance, frequency change, and travel terms from each individual trajectory.
Here Δx is the change in the SNV frequency vector over time, γ specifies the width of a Gaussian prior probability distribution for the selection coefficients si, and I is the identity matrix. Cint is the covariance matrix of SNV frequencies integrated over time, and accounts for competition between variants as well as the speed of growth for different viral lineages (Supplementary Information). τint accounts for travel and is given explicitly in equation (S2). Data from multiple outbreaks can be combined by summing contributions to the integrated covariance, frequency change, and travel terms from each individual trajectory.
Validation in simulations
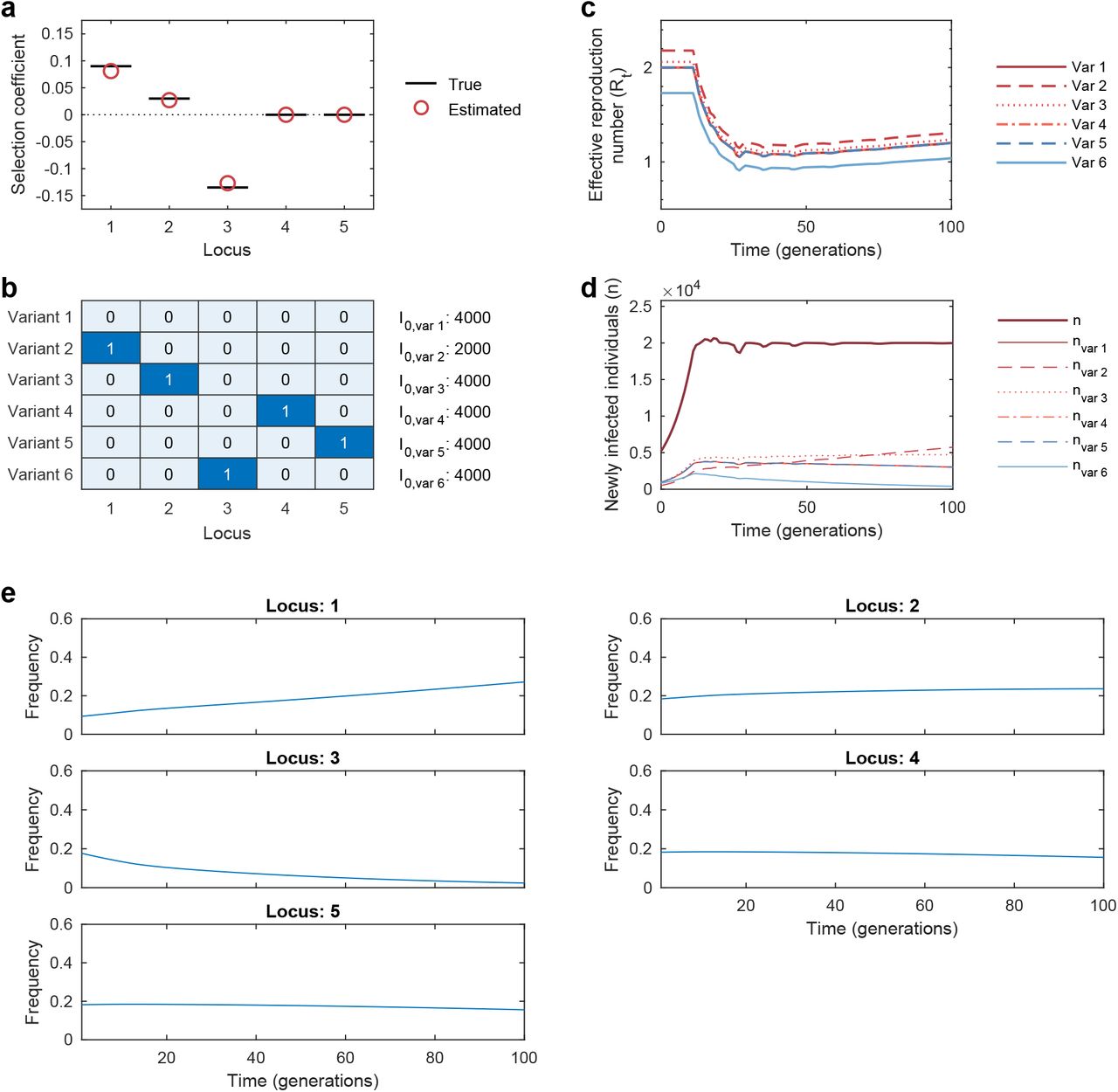
To test our ability to reliably infer selection, we analyzed simulation data using a wide range of parameters. We found that inference is accurate even without abundant data, especially when we combine information from multiple outbreaks (Fig. 1, Supplementary Fig. 1). Because we model the evolution of relative frequencies of different variants, accurate inference of selection does not require the knowledge of difficult-to-estimate parameters such as the current number of infected individuals or the effective reproduction number (Methods). Selection can be accurately inferred not only when the population evolves according to the superspreading model, but also if it evolves according to a classical multi-variant SIR model (Supplementary Figs. 2-3). This indicates that selection can be accurately estimated for a broad range of epidemiological dynamics.
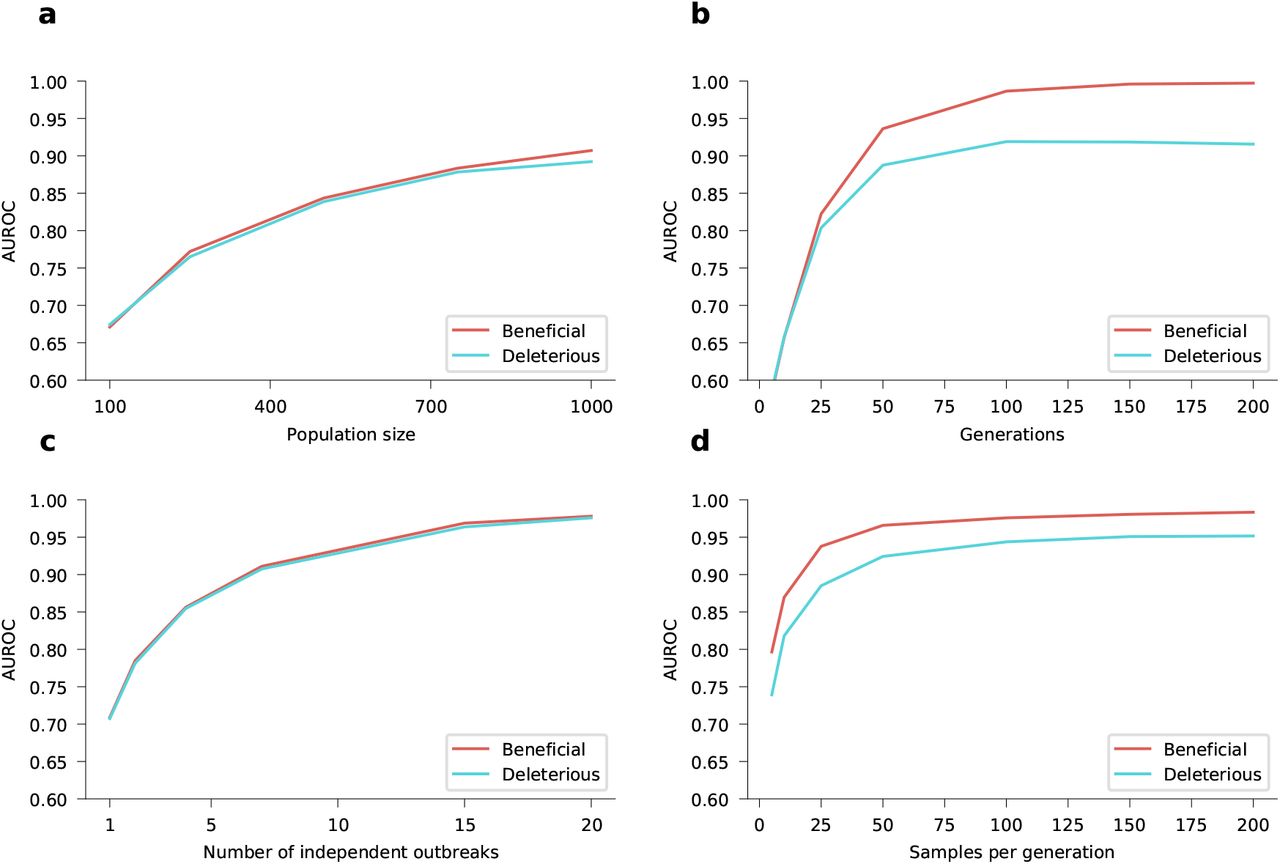
How the AUROC scores for both beneficial SNVs (in red) and deleterious SNVs (in blue) depends upon the different model parameters. a, Inference accuracy for different values of newly-infected population size. The parameters used are 10 simulations each with 50 sampled genomes per generation for 25 generations. b, Inference accuracy for different numbers of generations (serial intervals). Data is from a single simulation with 25 samples per generation and a newly-infected population size of 10,000. c, Inference accuracy for different numbers of independent outbreaks (simulations). The parameters used are 50 samples per generation for 10 generations and a newly-infected population size of 10,000. d, Inference accuracy for different values of samples per generations. Data is from a single simulation with 50 generations with a newly-infected population size of 10,000. The initial population is a mixture of two variants with beneficial SNVs (s = 0.03), two with neutral SNVs (s = 0), and two with deleterious SNVs (s = −0.03). Dispersion parameter k is fixed at 0.1. This is the same initial population composition as described in Fig. 1. All AUROC scores are calculated by averaging over 1,000 replicate simulations.

Here the simulation parameters are such that the number of newly infected individuals increases over time and then remains almost constant. a, True and estimated selection coefficients. We observe that the estimates are quite accurate. b, Variants present in the population along with the number of individuals infected by each variant at the initial time. c, Plot of effective reproduction number (Rt) of each variant over time. In this simulation we adjust the value of Rt by adaptively updating transmission rate β of each variant such that the total number of newly-infected individuals at each time remains almost constant. d, Plot of individuals newly-infected by each variant along with the total number of newly-infected individuals. e, Mutant frequency trajectories observed at each locus. Simulation parameters: Recovery rate for all variants is assumed to be the same, i.e., ra = r = 0.12, population size N = 10, 000, 000, the effective reproduction number Rt of the WT variant is initialized to a value of 2 while Rt for other variants is calculated from (11).

Here the simulation parameters are such that the number of newly-infected individuals follow the typical SIR dynamics. a, True and estimated selection coefficients. We observe that the estimates are quite accurate. b, Variants present in the population along with the number of individuals infected by each variant at the initial time. c, Plot of effective reproduction number (Rt) of each variant over time. In this simulation we adjust the value of Rt of each variant such that the total number of newly-infected individuals at each time follows typical SIR dynamics. d, Plot of individuals newly-infected by each variant along with the total number of newly-infected individuals. e, Mutant frequency trajectories observed at each locus. Simulation parameters: Recovery rate for all variants is assumed to be the same, i.e., ra = r = 1, population size N = 10, 000, 000, the transmission rate is adapted such that system follows the typical SIR dynamics.
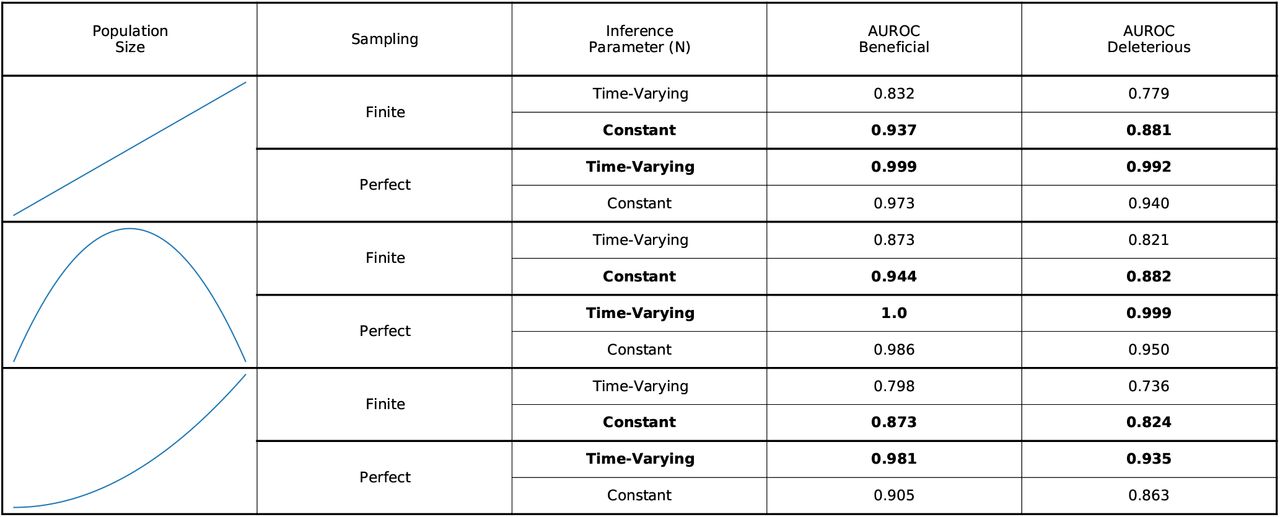
The ability of the model to distinguish beneficial and deleterious SNVs, as measured by the AUROC score, depending on whether the sampling is perfect or finite and whether constant arbitrary parameters or the true time-varying parameters are used for the population size n in the inference. Both simulations use constant values of k = 0.01 and R = 1. The results are similar but less dramatic if the correct time-varying values are used for k or R as well. Results are shown for different population trajectories and are consistent regardless of the trajectory. Rows that yield better inference are marked by bold text. If the sampling is finite, then it is better to use constant parameters; if the sampling is perfect, then it is better to use the real time-varying parameters. The initial population is a mixture of two variants with beneficial SNVs (s = 0.03), two with neutral SNVs (s = 0), and two with deleterious SNVs (s = −0.03), which is the same as that used in Fig. 1. Simulations are run for 50 simulations with 25 samples in each generation, and AUROC scores are averaged over 1,000 replicate simulations.

The number of genomes per day in the regions that are used for inference.

a-b, Comparison of inferred coefficients when the number of days at the beginning and end of the time-series are used in order to calculate the frequency changes. Inferred coefficients are largely robust to these changes c-d, Comparison of inferred coefficients for different frequency cutoffs. Including more or less sites does not alter the order of inferred coefficients. e-h, Comparison of inferred coefficients for different values of the regularization. Altering the regularization value has little effect upon the distribution of inferred selection coefficients, and selection coefficients for different values of the regularization are highly correlated.
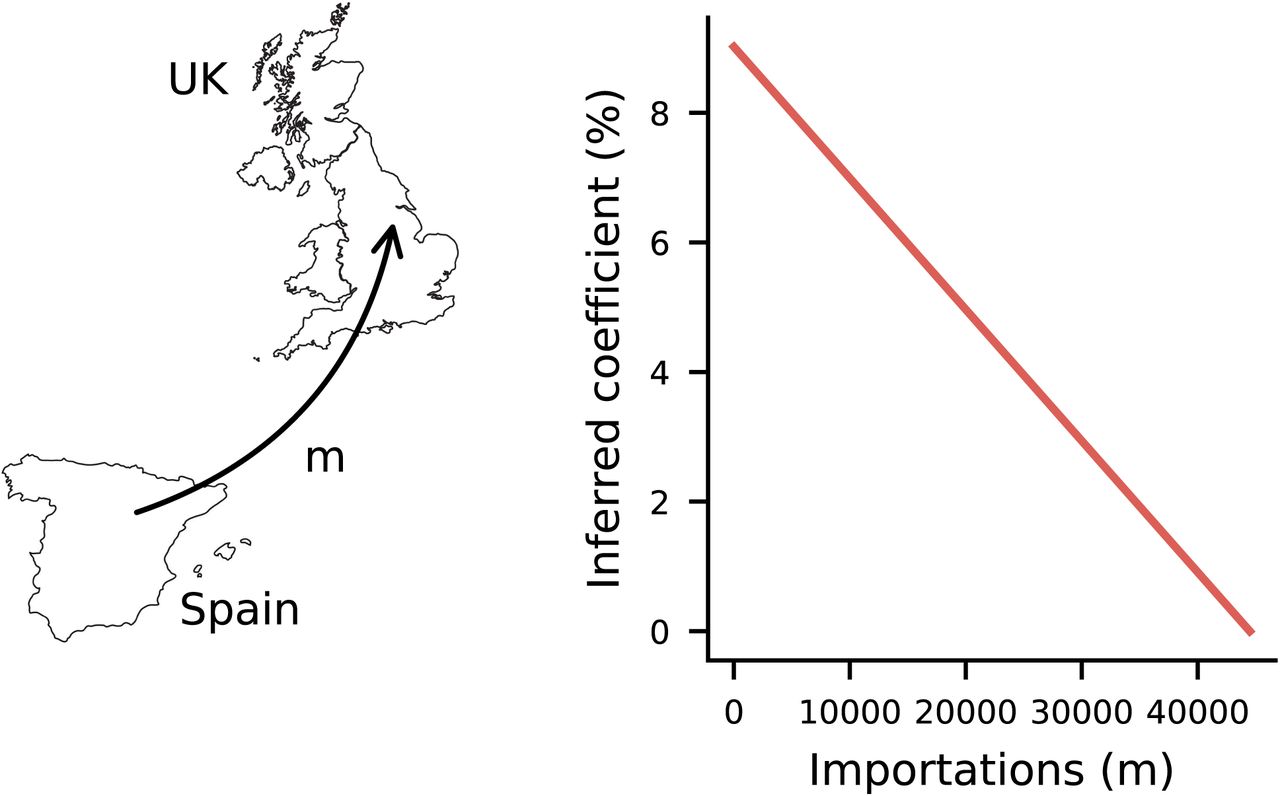
A plot of the inferred coefficient for the 20E (EU1) variant versus the total number of importations of this variant into the United Kingdom. The number of importations must be very large for the inferred coefficient to be small, indicating that travel of individuals infected with the variant 20E (EU1) is unlikely to fully explain the apparent fitness benefit provided by the group of SNVs.

The distribution of inferred selection coefficients for a neutral SNV when individuals who are infected with the variant travel into a regional outbreak. Simulation parameters: n = 10, 000 and k = 0.1. The number of newly infected individuals per serial interval rises rapidly from 6,000 to around 10,000 and stays nearly constant thereafter. The initial population is a mixture of two variants with beneficial SNVs (s = 0.03), two with neutral SNVs (s = 0), and two with deleterious SNVs (s = −0.03). This is the same initial population composition as in Supplementary Figure 1. 25 individuals infected with a variant containing a single neutral mutation travel into the population per generation for 100 generations. The distribution of inferred coefficients for the neutral SNV including travel (blue) and not including travel (red) is shown over 1,000 replicate simulations.

Simulated epidemiological dynamics beginning with a mixed population containing variants with beneficial, neutral, and deleterious mutations. a, Selection coefficients for individual SNVs, shown as mean values ± one theoretical s.d., can be accurately inferred from stochastic dynamics in a typical simulation (Methods). b, Extensive tests on 1,000 replicate simulations with identical parameters show that inferred selection coefficients are centered around their true values. Deleterious coefficients are slightly more challenging to accurately infer due to their low frequencies in data. Simulation parameters. The initial population is a mixture of two variants with beneficial SNVs (s = 0.03), two with neutral SNVs (s = 0), and two with deleterious SNVs (s = −0.03). The number of newly infected individuals per serial interval rises rapidly from 6,000 to around 10,000 and stays nearly constant thereafter. Dispersion parameter k is fixed at 0.1.
Global patterns of selection in SARS-CoV-2
We studied the evolutionary history of SARS-CoV-2 using genomic data from GISAID17 as of August 6th, 2021. We separated data by region and estimated selection coefficients jointly over all regions (Methods). After filtering regions with low or infrequent coverage, our analysis included more than 1.6 million SARS-CoV-2 sequences from 87 different regions, containing 13,189 nonsynonymous SNVs observed at nontrivial frequencies.
Our analysis revealed that, while the great majority of SNVs were nearly neutral, a few dramatically increased viral transmission (Fig. 2a, Table 1). We observe clusters of SNVs with strong effects on transmission along the SARS-CoV-2 genome (Fig. 2b). The highest density of SNVs that increase transmission is in Spike, especially in the S1 subunit. Of the top 20 mutations that we infer to be most strongly selected, 10 are in Spike (Table 1). However, clusters of SNVs with a strong selective advantage are also found in other proteins, especially in N, M, ORF3a, and NSP6.
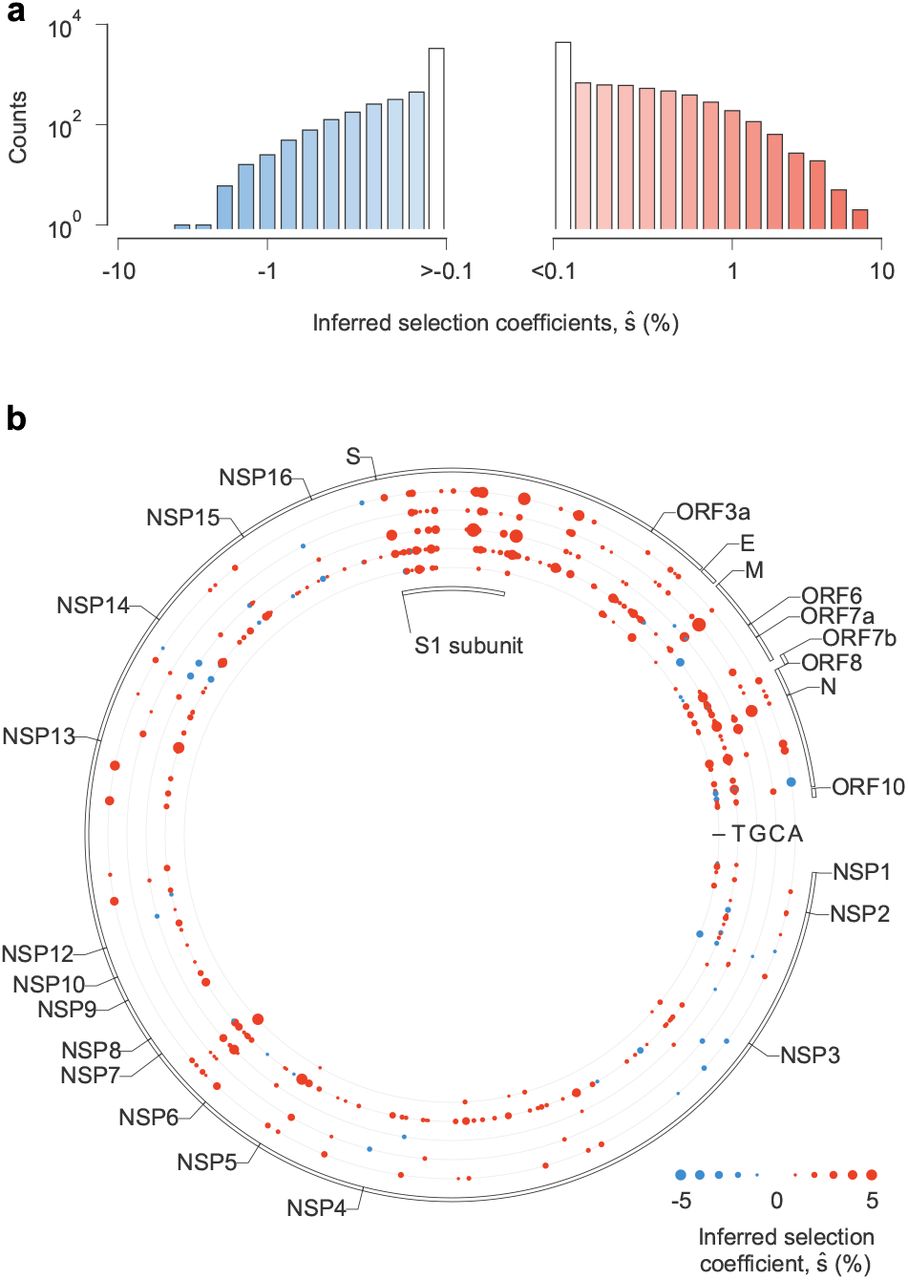
a, The vast majority of the 13,189 nonsynonymous SNVs included in our study are inferred to have negligible effects on transmission (that is, ŝ close to zero). However, a few SNVs have strong effects, as evidenced by a large value of ŝ. b, Patterns of selection across the SARS-CoV-2 genome. Beneficial SNVs often cluster together in the genome. Clustering is especially apparent for the S1 subunit of Spike, where many SNVs that are inferred to have the largest effects on transmission are located.
Table of most highly selected amino acid substitutions across the SARS-CoV-2 genome. * represents the cases where phenotypic effect of an amino acid variant has not been reported explicitly in the literature. Instead, it is either based on the function of the encompassing gene, for a mutation to a different amino acid or deletion at the same position, or for a mutation at a neighboring position. # all three mutations appear together; † each individual mutation leads to the same amino acid mutation; RBM = receptor binding motif; NTD= N-terminal domain; FCS = S1/S2 furin cleavage site; HR1 = heptad repeat 1; nAbs = neutralizing antibodies.
Table of selection coefficients for groups of amino acid mutations. Mutations that contribute most strongly to selection are listed first. The selection coefficient for a variant is calculated as the sum of the selection coefficients for the individual mutations that the variant contains.
Mutations inferred to substantially increase transmission
The top 50 mutations inferred to increase SARS-CoV-2 transmission the most are given in Table 1. Experimental evidence exists to directly or indirectly support many of these inferences. For clarity, we will reference mutations at the amino acid level rather than the underlying SNVs, which are also given in Table 1. Spike mutations L452R and P681R/H comprise three of the top four mutations, and all have demonstrated functional effects that could increase transmission4,18–21. Similarly, Spike mutations such as E484K (ŝ = 5.2%, ranked 9th) and S477N (ŝ = 4.4%, ranked 15th) appear prominently in our analysis. E484K has been shown to increase resistance to antibodies22, and both E484K and S477N were rapidly selected for increased ACE2 receptor binding during in vitro evolution23. Four Spike N-terminal domain (NTD) mutations/deletions (L18F, Δ141-142, and D253G) are also strongly selected. These lie in the antigenic supersite where mutations have been shown to decrease the neutralization potency of NTD-specific monoclonal antibodies24. Spike mutations D614G (ŝ = 2.9%, ranked 52nd) and N501Y (ŝ = 2.9%, ranked 58th) fall just outside the top 50 mutations in Table 1. D614G has been shown to increase binding affinity to the ACE2 receptor, thus increasing viral load and likely contributing to increased transmission7,25. Similarly, there is evidence that N501Y increases ACE2 binding affinity as well as transmission of infection26.
Research on viral transmission has naturally focused on Spike because of its role in viral entry and as a target of neutralizing antibodies. However, our analysis also reveals strongly selected mutations outside of Spike. These include the Nucleocapsid mutations D3L and R203M. D3L (ŝ= 6%, ranked 5th) has been reported to increase production of a non-canonical subgenomic RNA that encodes for ORF9b (ref.27), an interferon suppressing gene that can aid innate immune evasion and thereby increase transmission28. R203M (ŝ = 4.6%, ranked 13th), present in the linker region of Nucleocapsid, has been shown to enhance viral RNA replication, delivery, and packaging, which may increase transmission29. The Nucleocapsid mutation T205I (ŝ = 3.3%, ranked 40th) improves RNA delivery and expression to a lesser degree29. A Nucleocapsid mutation S202N (ŝ = 3.3, ranked 39th) is also inferred to be strongly selected. While the effect of this specific mutation is unknown, a mutation to Arginine at the same residue (S202R, which we infer to be more moderately beneficial, ŝ = 1.7%) has been reported to increase replication, RNA delivery, and packaging29. Other strongly selected mutations outside of Spike include the Membrane I82T mutation (ŝ= 7.5%, ranked 2nd) and NSP6 deletions Δ106-108 (ŝ = 5.4%, 4.2%, and 3.1%, ranked 7th, 23rd, and 43rd). Similar examples may provide good targets for future studies of the functional effects of non-Spike mutations.
Estimates of selection for major SARS-CoV-2 variants
We estimated the net increase in viral transmission relative to the Wuhan-Hu-1 reference sequence for well-known SARS-CoV-2 variants by adding contributions from the individual variant-defining SNVs (Fig. 3, see Methods). Because our model uses global data and infers the transmission effects of individual SNVs, variants can be compared to one another directly even if they arose on different genetic backgrounds, or if they appeared in different regions or at different times. This also allows us to infer substantially increased transmission for variants such as Gamma, Beta, Lambda, and Epsilon, which never achieved the level of global dominance exhibited by Alpha and Delta (Fig. 3). For reference, we show the selection coefficient inferred for the cluster of mutations including the Spike mutation D614G that fixed early in the pandemic.

Frequencies of major variants and their total inferred selection coefficients, defined relative to the Wuhan-Hu-1 reference sequence. Selection coefficients for variants with multiple SNVs are obtained by summing the effects of all variant-defining SNVs (Methods). Because our method uses global data and accounts for competition between variants, we infer large transmission advantages even for variants such as Gamma, Beta, Lambda, and Epsilon, which never achieved the same level of global dominance as variants such as Alpha and Delta.
Our findings are consistent with past estimates that have shown a substantial transmission advantage for Alpha and Delta relative to other lineages30–32. However, past estimates have varied substantially depending on the data source and method of inference. For example, in different analyses Delta has been inferred to have an advantage of between 34% and 97% relative to other lineages30–32. Similarly, Alpha has been estimated to increase transmission by 29% to 90% relative to pre-existing lineages in different regions5,11,12,31,33. One advantage of our approach is that it can infer selection coefficients that best explain the growth or decline of variants across many regions, allowing selection for different variants to be compared on even footing.
In November 2021, a new variant, Omicron, was detected in South Africa. The data that we consider only extends to August 6th, 2021, and thus Omicron is not present in our data set. However, because this variant bears SNVs observed in other lineages, we can provide a preliminary estimate of its transmission advantage. Even without considering the effects of 17 Omicron SNVs (out of 96 total) that were not previously observed in data, the total selection coefficient for Omicron is ŵ = 55.2%, which surpasses Alpha. While more data will be necessary to fully assess the transmission advantage of this variant, our model suggests that Omicron is highly transmissible, supporting its designation as a variant of concern (VOC) by WHO34.
Effects of travel and competition on inferred coefficients
In addition to differences in transmissibility, variant frequencies are affected by the travel of infected individuals between regions and competition between variants. Multiple introductions of a new variant into a region can increase its local frequency even if the variant has no transmission advantage35,36. This could, for example, make a neutral variant appear beneficial. Conversely, variants that transmit more effectively than ancestral SARS-CoV-2 can still be outcompeted by other, more transmissible variants, causing them to decline. In population genetics, this is referred to as clonal interference37,38.
We studied the history of Nextstrain lineage 20E (EU1) as an example to investigate the influence of travel and inter-lineage competition on inferred changes in transmission. A detailed analysis showed how 20E (EU1) spread from Spain to other regions in Europe35. There it was estimated that 20E (EU1) was introduced into the UK roughly 380 times during the summer of 2020. Assuming no travel between the UK and other regions, we infer a total selection coefficient for novel 20E (EU1) SNVs of 15.6% using UK data gathered from the beginning of the pandemic through May 1st, 2021, when 20E (EU1) had died out locally. Including 380 importations into the UK during the summer of 2020, our inferred selection coefficient is only slightly reduced to 15.4% (Methods). Around 37,000 importations would be necessary during this time for 20E (EU1) to be inferred to be completely neutral. Thus, while travel was crucial for the early spread of 20E (EU1) in the UK and across Europe, its subsequent growth ultimately dominates estimates of its effects on transmission.
Competition between Alpha and 20E (EU1), which was rising in the UK before the emergence of Alpha, provides a clear example of clonal interference in SARS-CoV-2. Interference can be readily observed in their frequency trajectories in the UK (Fig. 4a). Changes in frequency for the two variants have a Pearson correlation of −0.73.
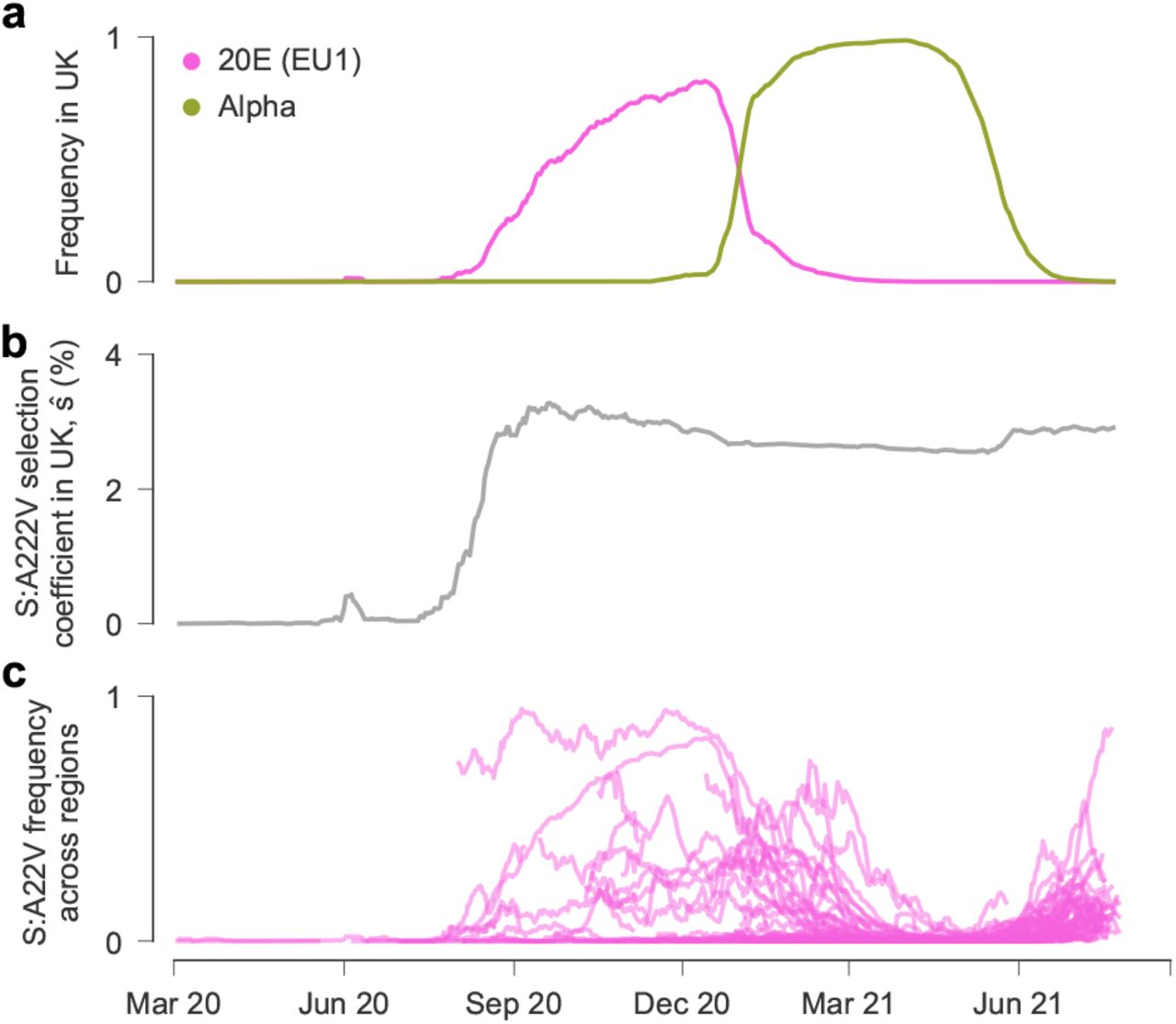
a, The 20E (EU1) lineage, bearing the Spike mutation A222V, is outcompeted by Alpha in the UK. b, Because our model accounts for competition between variants, we nonetheless infer a transmission benefit for A222V, as well as other 20E (EU1) mutations, that persists even as 20E (EU1) dies out in the UK. c, Globally, S:A222V has arisen on different sequence backgrounds and increased in frequency since June 2021, consistent with a transmission advantage for this mutation.
To measure the influence of competition on estimates of transmission, we inferred selection coefficients for variants circulating in the UK with all nucleotide changes present in the Alpha variant reverted to the Wuhan-Hu-1 reference sequence. This provides an estimate for the 20E (EU1) selection coefficient that ignores competition with Alpha. Using all of the data, 20E (EU1) is inferred to have a significant transmission advantage relative to the previously dominant variant B.1 (ŵ20E(EU1) − ŵB.1 = 10.2%). However, when competition with Alpha is ignored, 20E (EU1) is inferred to be mildly deleterious (ŵ20E(EU1) − ŵB.1 = − 1.7%). Thus, ignoring competition between variants would lead to a dramatic and likely incorrect change in inferred selection.
The re-emergence of the Spike mutation A222V, which appeared within the 20E (EU1) variant, further supports a transmission advantage. Using global data, we infer A222V to significantly increase transmission (ŝ = 3.8%, ranked 29th, see Table 1). In our analysis, A222V contributes far more than any other mutation to the increased transmission of 20E (EU1) relative to contemporary variants. Focusing on data from the UK alone, the inferred benefit of A222V remains roughly constant even as 20E (EU1) is outcompeted by Alpha (Fig. 4b). Later, A222V arises on other sequence backgrounds and steadily increases in frequency (Fig. 4c), consistent with the increase in transmission inferred by our model.
Our analysis therefore suggests that 20E (EU1) possessed a modest but real transmission advantage relative to contemporary SARS-CoV-2 sequences. This finding is consistent with analysis in ref.35, where a model of 20E (EU1) spread due to travel alone underestimated the observed frequency of the variant by 1-to 12-fold in all regions. More generally, our analysis suggests that very large inferred selection coefficients for variants or SNVs in our model are unlikely to be explained by travel alone.
Rapid detection of variants with enhanced transmission
Rapidly identifying variants with increased transmission is important to inform public health efforts to limit viral spread. However, the inherent stochasticity of infection and of genomic surveillance data collection makes accurate inferences difficult. For example, neutral or modestly deleterious variants may initially appear to be beneficial due to a transient rise in frequency despite having no selective advantage.
To quantify how fluctuations affect estimates of selection for neutral variants, we first identified all variants (including both SNVs and collections of SNVs that are strongly linked to one another) that are inferred to have selection coefficients with magnitude less than 1% using all of the data. We then calculated the selection coefficients that would have been inferred for the SNVs or variants at all earlier time points and in all regions after they were first observed in the data. This “null” distribution (Fig. 5a) quantifies fluctuations in inferred selection coefficients for nearly-neutral variants due to stochasticity in viral spread and sampling. Variants with selection coefficients larger than any in the null distribution could then be expected with high confidence to have some transmission advantage.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
a, The null distribution of inferred selection coefficients for neutral variants over all intermediate times and across all regions. Neutral variants are defined as (collections of) SNVs that are inferred to have total selection coefficients |w| < 1% using global data. b, Frequency of Alpha in London and its inferred selection coefficient over time. The inferred coefficient exceeds the largest one in the null distribution on November 9th, 2020. c, Frequency of Delta in the UK and the inferred selection coefficient for novel Delta SNVs over time. The inferred coefficient exceeds the null on March 31st, 2021.
To assess our ability to rapidly detect variants with a transmission advantage, we studied the rise of the Alpha variant in the London area. Using the above criterion, we identify Alpha as very likely to increase transmission using sequence data that was collected on or before November 9th, 2020 (Fig. 5b). This is roughly three weeks before Public Health England labeled Alpha as a variant of interest (VOI)39, and more than a month before it was classified as a VOC40. At this time the frequency of Alpha in London was around 1%.
A similar analysis also shows that our model rapidly infers increased transmission for the Delta variant. Using data from the UK, we reliably infer Delta to increase transmission by March 31st, 2021 (Fig. 5c). Delta was classified as a VOI on April 4th, 2021 and as a VOC more than one month later on May 6th, 202141. At the time that we detected increased transmission for Delta, its frequency was still low (< 1%) in the UK. Collectively, these results demonstrate our ability to rapidly identify variants with higher transmission, even when they represent a small fraction of all infections in a region.
Discussion
Quantifying the effects of mutations on viral transmission is an important but challenging problem. To overcome limitations of current methods, we developed a flexible, SIR-based epidemiological model that provides analytical estimates for the transmission effects of SNVs from genomic surveillance data. Applying our model to SARS-CoV-2 data, we identified SNVs that substantially increase viral transmission, including both experimentally-validated Spike mutations and other, less-studied mutations that may be promising targets for future investigation. We further explored the effects of travel and competition between variants on inferred changes in transmission, using the history of 20E (EU1) as an example. Importantly, we found that our model is sensitive enough to detect substantial transmission advantages for variants such as Alpha and Delta even when they comprised only a small fraction of the total number of infections in a region, thus providing an “early warning” for more transmissible variants.
Further monitoring will be important to identify and characterize new variants as they arise. The Omicron variant that was recently detected in South Africa provides one such example. While the data in our study only extends to August 6th, 2021, we would estimate a selection coefficient of 55.2% for Omicron based on the mutations that it shares with previous variants alone.
While our study has focused on SARS-CoV-2, the epidemiological model that we have developed is very general. The same methodology could be applied to study the transmission of other pathogens such as influenza. Combined with thorough genomic surveillance data, our model provides a powerful method for rapidly identifying more transmissible viral lineages and quantifying the contributions of individual mutations to changes in transmission.
Data Availability
Sequence data and metadata used in this study are available from GISAID. Processed data and code are available in the linked GitHub repository.
AUTHOR CONTRIBUTIONS
All authors contributed to methods development, data analysis, interpretation of results, and writing the paper. B.L. and J.P.B. led theoretical analyses. M.S.S. led SIR model simulations. B.L. led branching process simulations. J.P.B. conceptualized and supervised the project.
Methods
Epidemiological model
We use a discrete time branching process to model the spread of infection. Individuals can be infected by any one of M viral variants, which are represented by genetic sequences g = {g1, g2, …, gL} of length L. For simplicity, we will first assume that alleles at each site i in the genetic sequence for variant a are either equal to the “wild-type” or reference 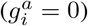 or mutants
or mutants  . Later we will relax this assumption to consider genetic sequences with 5 possible states at each site (4 nucleotides or a gap). We call na(t) the number of individuals infected by variant a at time t. To account for super-spreading, the number of newly infected individuals at time t + 1 follows a negative binomial distribution42, P (na(t + 1)|na(t), k, Ra) = PNB (r, p), where r = nak, p = k/(k + Ra), and Ra = R(1 + wa). Here r and p are the negative binomial distribution parameters, k is the dispersion, R is the average effective reproductive number, and wa encodes the variant dependence of the infectivity. Here R is also an implicit function of time, representing change in the number of susceptible and recovered individuals as well as the effects of public health interventions or changes in behavior that modify viral transmission.
. Later we will relax this assumption to consider genetic sequences with 5 possible states at each site (4 nucleotides or a gap). We call na(t) the number of individuals infected by variant a at time t. To account for super-spreading, the number of newly infected individuals at time t + 1 follows a negative binomial distribution42, P (na(t + 1)|na(t), k, Ra) = PNB (r, p), where r = nak, p = k/(k + Ra), and Ra = R(1 + wa). Here r and p are the negative binomial distribution parameters, k is the dispersion, R is the average effective reproductive number, and wa encodes the variant dependence of the infectivity. Here R is also an implicit function of time, representing change in the number of susceptible and recovered individuals as well as the effects of public health interventions or changes in behavior that modify viral transmission.
To incorporate travel, na is the sum of the locally infected population and the net flux of infected individuals into the region, na = na,local + na,inflow − na,outflow ≡ na,local + δna. Defining the frequency of variant a as ya = na/Σb nb, the probability that the frequency vector is y(t + 1) = {y1(t + 1), y2(t + 1),…} given the initial frequency vector y(0), is
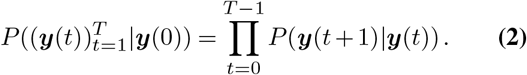
Derivation of the estimator
Because (2) is difficult to work with directly, we introduce a “diffusion approximation” where we assume that the total number of infected individuals is large and the effects of mutations on transmission are small. Similar approximations have been widely used in population genetics43–45. Under these assumptions, the probability distribution for the variant frequencies satisfies a Fokker-Planck equation with terms derived from the first and second moments of the frequency changes ya(t + 1) − ya(t) under the negative binomial distributions above.
However, the genotype space is high-dimensional (dimension 2L, with either a mutant or wild-type allele at each site) and undersampled, making inference of selection for genotypes extremely challenging. To simplify the inference problem, we assume that selection is additive, so the total selection coefficient wa for a variant a is the sum of selection coefficients si for mutant alleles at each site i:
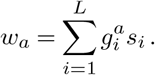 We can then derive a Fokker-Planck expression for the dynamics of mutant allele frequencies
We can then derive a Fokker-Planck expression for the dynamics of mutant allele frequencies
 At the allele level, the Fokker-Planck equation has a drift vector given by
At the allele level, the Fokker-Planck equation has a drift vector given by
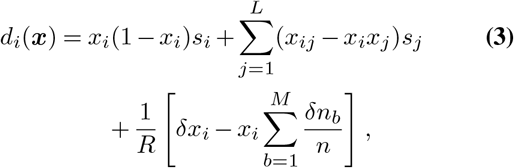 and a diffusion matrix
and a diffusion matrix
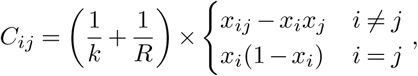 where xij is the frequency of infected individuals that have mutant alleles at both site i and site j at time t, and δxi is the change in the frequency due to individuals traveling. Here
where xij is the frequency of infected individuals that have mutant alleles at both site i and site j at time t, and δxi is the change in the frequency due to individuals traveling. Here 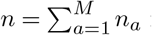 is the total number of individuals infected by all variants. If li = li,inflow − li,outflow is the number of people traveling into a region minus that traveling out of it who are infected with a variant that has a mutant allele at site i, then δxi = li/n.
is the total number of individuals infected by all variants. If li = li,inflow − li,outflow is the number of people traveling into a region minus that traveling out of it who are infected with a variant that has a mutant allele at site i, then δxi = li/n.
The Fokker-Planck equation can then be used to derive a path integral, which expresses the probability of an entire evolutionary history or “path” (i.e., frequencies of genetic variants over time,  . The path integral is
. The path integral is
 The path integral quantifies the probability density for paths of mutant allele frequencies in the evolutionary history of the pathogen. We can then use Bayesian inference to find the maximum a posteriori estimate for the selection coefficients given the frequencies, the infected population size, the parameters R and k, and the composition of the population traveling into or out of the regional outbreak. The posterior probability of the selection coefficients is
The path integral quantifies the probability density for paths of mutant allele frequencies in the evolutionary history of the pathogen. We can then use Bayesian inference to find the maximum a posteriori estimate for the selection coefficients given the frequencies, the infected population size, the parameters R and k, and the composition of the population traveling into or out of the regional outbreak. The posterior probability of the selection coefficients is
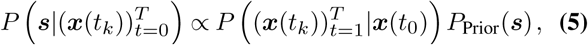 where
where  is the probability of a path given by (4) and the PPrior(s) is a Gaussian prior probability for the selection coefficients with zero mean and covariance matrix σ2I. Here, I is the identity matrix and σ2 is the variance of the prior. The selection coefficients that maximize (5) are
is the probability of a path given by (4) and the PPrior(s) is a Gaussian prior probability for the selection coefficients with zero mean and covariance matrix σ2I. Here, I is the identity matrix and σ2 is the variance of the prior. The selection coefficients that maximize (5) are
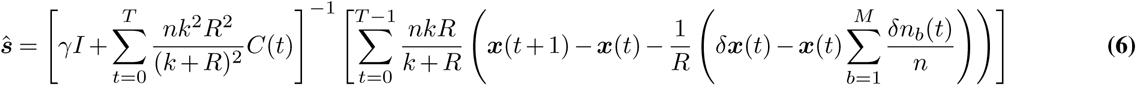 where the parameters k, R, and n are implicitly functions of t.
where the parameters k, R, and n are implicitly functions of t.
There are two interesting limiting forms of the estimator. First, we define the new matrix 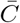 whose entries are
whose entries are
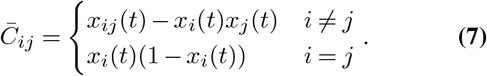 In the limit that k → ∞, the negative binomial distribution for new infections becomes a Poisson distribution with rate λ = R. In this special case, the model is equivalent to the Wright-Fisher model from population genetics. The estimator reduces to
In the limit that k → ∞, the negative binomial distribution for new infections becomes a Poisson distribution with rate λ = R. In this special case, the model is equivalent to the Wright-Fisher model from population genetics. The estimator reduces to
 where we have dropped the migration term for simplicity.
where we have dropped the migration term for simplicity.
The opposite limit k → 0 corresponds to a distribution for new infections with extremely heavy tails, i.e., one where super-spreading is dominant. In this case the drift in (3), which quantifies expected frequency changes due to selection and travel, is unchanged. However, the diffusion matrix, which encodes linkage as well as the changes in frequency that are due to the stochastic nature of infection transmission, diverges. In this case, diffusion dominates the process entirely.
Simplifying the estimator and robustness to incomplete knowledge of time-varying parameters
In practice, parameters appearing in (6), such as the infected population size n, the dispersion k, and the mean reproductive number R, are likely time-varying. While such time dependencies are accommodated by our model, they can be challenging to reliably estimate from data. However, we generally do not require full knowledge of these time-dependent parameters to accurately estimate selection.
In fact, due to finite sampling noise, estimates of selection produced by assuming constant (and incorrect) parameters are more accurate than estimates that use the true time-varying parameters (Supplementary Fig. 4). The naive estimator in (6) implies that time points or regions with larger R, n, or k should be weighted more heavily in the estimate. However, frequency information is always inaccurate due to noise from finite sampling, so weighing some time points or regions significantly more than others based upon the parameters alone means that undue weight is given to the uncertain information available from these times and regions.
For this reason, we assume parameters that are spatially and temporally constant in all of the following analysis, except when considering travel, as discussed below. This allows the estimator to be simplified substantially. If we assume constant parameters and scale the regularization γ by the prefactor in the numerator in (6), the parameter dependence in the numerator and the denominator is almost the same and largely cancels out. With the same definition of the matrix 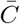 as above, and additionally defining
as above, and additionally defining 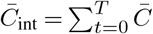 , the simplified estimator is given by
, the simplified estimator is given by
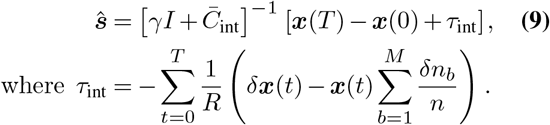 This form of the estimator has significant advantages over (6). The most important is that, if the travel term is dropped, the difficult-to-estimate parameters R, k, and n are no longer required. For methods of inferring these parameters as well as discussions about the difficulty of inferring them, see refs.46–55.
This form of the estimator has significant advantages over (6). The most important is that, if the travel term is dropped, the difficult-to-estimate parameters R, k, and n are no longer required. For methods of inferring these parameters as well as discussions about the difficulty of inferring them, see refs.46–55.
Extension to multiple regions and multiple SNVs at each site
The model can easily account for outbreaks in multiple regions or outbreaks at different times. If the probability of the evolutionary path in each region is independent, which is the case if there is no travel between regions, then the probability of all of the evolutionary paths in all of the regions is simply the product of the probabilities of the paths in each region, given by (4). Bayesian inference can be applied in the same way as before, resulting in the estimator
 where Q is the number of regions, tr is the time in region r, Tr is the final time in region r, tr,0 is the initial time in region r, xr is the frequency in region r, and
where Q is the number of regions, tr is the time in region r, Tr is the final time in region r, tr,0 is the initial time in region r, xr is the frequency in region r, and  is the scaled integrated covariance matrix in region r given by integrating (7) over time. The estimator can further be extended to allow for multiple different nucleotides at each site by simply letting each different nucleotide have its own entry in the frequency vector xi. If there are J mutations at each site this results in a frequency vector of length LJ, and a covariance matrix of size LJ × LJ. By convention, reference sequence alleles have selection coefficients of zero, so the mutant allele selection coefficients at each site are normalized by subtracting the inferred coefficient for the reference allele.
is the scaled integrated covariance matrix in region r given by integrating (7) over time. The estimator can further be extended to allow for multiple different nucleotides at each site by simply letting each different nucleotide have its own entry in the frequency vector xi. If there are J mutations at each site this results in a frequency vector of length LJ, and a covariance matrix of size LJ × LJ. By convention, reference sequence alleles have selection coefficients of zero, so the mutant allele selection coefficients at each site are normalized by subtracting the inferred coefficient for the reference allele.
Branching process simulations
We implemented the superspreading branching process for the number of infected individuals in Python. We used a negative binomial distribution for the number of secondary infections caused by a group of individuals infected with the same pathogen variant. To test how finite sampling affects model estimates, we sampled ns genomes per time point to use for analysis. We computed the single and double mutant frequencies, xi and xij, respectively, from the sampled sequences and estimated the selection coefficients from these using (1), possibly extended to account for multiple outbreaks or multiple alleles at each locus as described above.
Susceptible-infected-recovered simulations
We simulated a multi-variant Susceptible-Infected-Recovered (SIR) model with M variants of a pathogen circulating in a population of N individuals, assuming total cross-immunity between variants. Mathematically, the SIR dynamics are expressed as
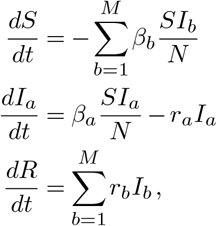 for a = 1, …, M, where Ia is the number of individuals infected by variant a, βa and ra are the transmission and recovery rate associated with the ath variant, and S and R are the total number of susceptible and recovered individuals, respectively. Each variant is represented by a binary sequence of length L, with 0 representing the wild-type (WT) allele and 1 representing the mutant allele. Considering the all-zero sequence as the reference with transmission rate βref, the transmission rate of variant a can be expressed as
for a = 1, …, M, where Ia is the number of individuals infected by variant a, βa and ra are the transmission and recovery rate associated with the ath variant, and S and R are the total number of susceptible and recovered individuals, respectively. Each variant is represented by a binary sequence of length L, with 0 representing the wild-type (WT) allele and 1 representing the mutant allele. Considering the all-zero sequence as the reference with transmission rate βref, the transmission rate of variant a can be expressed as  , with
, with 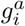 and si defined as above.
and si defined as above.
We further incorporate the effect of public health interventions and changing human behaviour on transmission by making the transmission rate a function of time, i.e., βref(t). As the number of susceptible individuals decreases, the effective transmission rate will decrease. The effective reproductive number of the ath variant at time t is
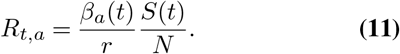 We used MATLAB to simulate the SIR model under a scenario where the number of newly-infected individuals continues to increase and then remains fixed (Supplementary Fig. 2), and a scenario where we fix ra = 1 and adapt the transmission rate over time such that the system follows the typical SIR dynamics (Supplementary Fig. 3). In the SIR model there is no superspreading, which corresponds to the limit k → ∞ in the branching process model described above. The estimator for the selection coefficients then reduces to a scaled version of (8),
We used MATLAB to simulate the SIR model under a scenario where the number of newly-infected individuals continues to increase and then remains fixed (Supplementary Fig. 2), and a scenario where we fix ra = 1 and adapt the transmission rate over time such that the system follows the typical SIR dynamics (Supplementary Fig. 3). In the SIR model there is no superspreading, which corresponds to the limit k → ∞ in the branching process model described above. The estimator for the selection coefficients then reduces to a scaled version of (8),
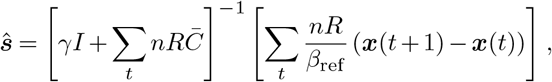 where n = n(t) is the number of newly-infected individuals at time t (and is different therefore from I), x is the frequency vector of newly infected individuals, and γ is the regularization. In both cases selection coefficients are accurately recovered.
where n = n(t) is the number of newly-infected individuals at time t (and is different therefore from I), x is the frequency vector of newly infected individuals, and γ is the regularization. In both cases selection coefficients are accurately recovered.
Regions and time-series for SARS-CoV-2 analysis
We used sequence alignments and metadata downloaded from GISAID (ref.56) on August 14th, 2021, which includes more than 3 million sequences. Ideally, we would like to divide this data into the smallest separate areas that have outbreaks that are largely independent of those in the surrounding regions, so as to avoid biases due to travel between regions or unequal sampling in different locations. However, this needs to be balanced with the limitations of the data, since regions with poor sampling could contribute more noise than signal. We therefore divided data into the smallest regions available in the metadata that are still large enough such that infections resulting from travel outside of the region are likely to be far less frequent than transmission within the region. This results in the inclusion of mostly separate countries in Europe and Asia and states in North America. Two exceptions to this are that we separate northern and southern California due to the geographical separation of population centers, and we separate Northern Ireland from the rest of the United Kingdom due to its geographical isolation.
To minimize the effects of sampling noise, we chose regions and time-series within these regions based on the following criteria:
In any period of 5 days within the time-series there are at least 20 total samples.
The number of days in the time-series is greater than 20.
The number of new infections per day is at least around 100.
The last criterion ensures that there are enough infected individuals that transmission is not driven overwhelmingly by stochasticity.
Our results are robust to reasonable variation in these parameters. Comparing the number of locations used and the sample sizes shown in Supplementary Fig. 5 in the data to those used in the simulations shown in Supplementary Fig. 1, we expect our inference to accurately distinguish beneficial, deleterious, and neutral SNVs from one another.
Data processing
We perform a number of preprocessing steps to ensure data quality. We first eliminated incomplete sequences with gaps at more than one third of the genome. We then removed sites from our analysis where gaps are observed at > 95% frequency, since these sites may represent very rare insertions or sequencing errors. We also removed sites in noncoding regions of the SARS-CoV-2 genome and ones where all observed SNVs are synonymous. We imputed ambiguous nucleotides with the nucleotide at the same site that occurs most frequently in other sequences from the same region.
For the remaining sites, we excluded rare SNVs whose frequency is never larger than 1% in any region and ones that are not observed at least 5 times. These sites, if included, are almost always inferred to have extremely small selection coefficients. Furthermore, since their frequencies are so small, their covariance with other sites is also small and is therefore unlikely to have a large effect on inference. We verified that different reasonable values for these cutoffs result in essentially identical selection coefficients (Supplementary Fig. 6).
Calculating frequency changes and covariances
To increase robustness to finite sampling, we integrated terms in (6) over time, assuming that frequencies and covariances are piecewise linear, rather than summing contributions from each time point57. To obtain better estimates of changes in SNV frequencies (the term x(T) − x(0) in (9)), we averaged x(T) as the frequencies in the window of the final 10 days and x(0) as the frequencies in the window of the first 10 days for each time-series and region. This smoothing is necessary especially in regions where sampling is sparse, where the number of genomes sampled on a particular day may be as small as 1 or 2. We confirmed that our results are robust to reasonable changes of this window size of 10 days (Supplementary Fig. 6).
We also normalized time in units of serial intervals or “generations” by dividing the integrated covariance matrix by 5, following results that the serial interval for SARS-CoV-2 is roughly 5 days58–60. This allows us to convert from units of time in days to generations, as in (9).
Calculating selection coefficients
After the above preprocessing (before eliminating synonymous SNVs) there remain 21, 050 SNVs observed at a frequency above 1% in at least one region and observed at least 5 times. We assume constant values for R, n, and k in all regions, and use (10) to estimate selection. When R, n, and k are constant, these terms can be effectively absorbed into the regularization γ.
We normalize selection coefficients such that the nucleotide for the Wuhan-Hu-1 reference sequence at each site has a selection coefficient of 0. To do this, we subtract the selection coefficient for the reference nucleotide from the inferred coefficient for each other allele at that site after all selection coefficients have been computed.
We used these estimates for the selection coefficients for nonsynonymous SNVs to estimate the corresponding selection coefficients for amino acid substitutions (Table 1). If there were multiple SNVs in a codon that result in the same amino acid variant, but are not strongly linked to one another, then the selection coefficient for the amino acid was calculated as the largest (in absolute value) of the SNVs. If there were multiple SNVs in the same codon that yield the same amino acid and these SNVs are strongly linked to one another, then the selection coefficient for the mutant amino acid was calculated as the sum of the selection coefficients for the SNVs.
We calculated selection coefficients for major variants by summing the individual nucleotide SNVs that define the variant, which follows from our assumption of additive fitness. SNVs for major variants were obtained by first finding groups of strongly linked SNVs that correspond to a variant, and then adding any other mutations given on https://covariants.org that were not identified by our linkage analysis.
We also computed selection coefficients for collections of strongly linked SNVs that may not be officially-designated variants. To determine sets of strongly linked SNVs, we considered the following statistics. If the number of genomes with a SNV at site i is called hi and the number of genomes with SNVs at both site i and site j is hij, then we say that two sites i and j are strongly linked if hij/hi and hij/hj are both greater than 80%. As for the major variants, we computed selection coefficients for sets of strongly linked SNVs by summing the contributions from individual SNVs. Selection coefficients for strongly linked SNVs were used to compute the “null” distribution that we use as a metric for early detection of variants with increased transmission.
Choice of regularization
In principle, the regularization strength γ is related to the width of the prior distribution for SNV selection coefficients. The regularization strength also plays a role in reducing noise in selection coefficient estimates due to finite sampling of viral sequences. This is especially important for SNVs that are observed only briefly in data, as they will have small integrated variances in the “denominator” of (6). Larger values of the regularization more strongly suppress noise, but they also shrink inferred selection coefficients towards zero.
We use a regularization strength of γ = 40 after absorbing factors of n, k, and R into γ (see (S4) in Supplementary Information). For much smaller values of γ, selection coefficient estimates are unstable due to sampling noise. However, inferred selection coefficients stabilize and become insensitive to the precise value of γ for γ ≳ 10 (Supplementary Fig. 6). Larger values of γ will result in selection coefficients with smaller absolute values, but for large enough γ the rank ordering of inferred selection coefficients is highly reliable. In summary, the coefficients that appear to be the most beneficial or deleterious remain this way regardless of reasonable choices for γ, though their precise values scales with the regularization strength.
Rapid detection of variants with increased transmission
To estimate how quickly we can detect a transmission advantage for a new SNV or variant, selection coefficients are calculated only in the specific region where the variant arose. Since inference is only done in a single region, SNVs that appear only briefly at low frequencies —and which therefore are unlikely to change transmission rate —only appear once, whereas in the global analysis such SNVs may appear at low frequencies in multiple regions. For this reason we use a lower regularization of 10 for regional analysis. The null distribution is calculated by first finding all variants (including one or more SNVs) that are inferred to have a selection coefficient of absolute value less than 1% using the joint inference over all regions. We then calculated the selection coefficients that would have been inferred for these variants at all earlier time points in each region after they were first observed in that region. We can then say with high confidence that a variant increases transmission once the inferred coefficient for that variant in a specific region surpasses any of the inferred coefficients in the null distribution.
Effects of travel on inferred selection
Travel of infected individuals can bias inferred selection coefficients by changing the frequency of variants in a region for reasons that are not due to increased or decreased transmission. To analyze the effect that travel has on the inferred selection coefficients, we focused on the United Kingdom, and especially on the variant 20E (EU1), because sampling the UK is excellent and because there exists a high-quality estimate for the number of importations of this variant61. In order to quantify the effect of travel, it is important to have an estimate for the number of newly infected individuals on each day since the effect due to travel depends on the number of cases that are imported relative to the number of local cases (see 6). We used statistics from the Institute for Health Metrics and Evaluation62 to estimate the number of newly infected individuals in the UK. For simplicity, we assumed a constant number of importations of 20E (EU1) per day starting on July 7th, 2020 (the first day a 20E (EU1) sequence was sampled in the UK) and continuing for 100 days. We then inferred the selection coefficients for many different numbers of importations. The results are shown in Supplementary Fig. 7, where we find that a very large number of importations is necessary for 20E (EU1) to be inferred to be neutral (ŵ = 0).
In our full data set, selection coefficients that are inferred to be close to zero may in fact be slightly beneficial or deleterious and are inferred incorrectly due to travel. However, given the degree of travel needed to substantially bias inferred selection demonstrated in Supplementary Fig. 7, travel is unlikely fully explain large inferred selection coefficients. This is especially true for variants observed in regions where travel restrictions reduced the number of infected individuals entering or leaving a region. In addition, the effects of travel are also muted when the number of local infections is large.
Data and code
Sets of processed data, computer code, and scripts that we have used in our analysis are available in the GitHub repository located at https://github.com/bartonlab/paper-SARS-CoV-2-transmission. This repository also contains Jupyter notebooks that can be run to reproduce the results presented here, using sequence data and metadata from GISAID.
Supplementary Information
1. Summary
Here we discuss three main topics. First, we give a detailed introduction of our epidemiological model as well as a derivation of the estimator (1). We then describe simulations of an outbreak and show that selection coefficients can be accurately recovered from simulation data even with relatively poor sampling. Finally, we discuss our analysis of SARS-CoV-2 evolution during the outbreak. We recover known results, and we show that inference is insensitive to a large variety of parameter choices.
2. Epidemiological model
2.1. Introduction
In epidemiology, the spread of infection can be modeled as a branching process where each infected individual (also referred to as a case) infects n additional individuals1. The distribution of n is often taken to be Poisson, but differences in the number of contacts with susceptible individuals, disease course within an individual, and other factors mean that the Poisson rate λ is not generally the same for all cases2. Below, we first follow ref.2 to explore families of distributions for the number of new cases per infected individual. Next, we extend these models to consider multiple variants of the pathogen that differ in their spreading efficiency. We seek to characterize how the distribution of pathogen variant frequencies is expected to change over time, and how such data can be used to estimate the relative spreading efficiency of different variants.
2.2. Distributions for the number of infected individuals
As noted above, the basic distribution of the number of new cases n caused by one case in a susceptible population is Poisson,
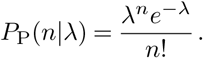 Typically we might take the Poisson rate λ to be R, the effective reproduction number, which is the expected number of cases directly caused by one case. In that case, the average number of cases following the Poisson distribution is
Typically we might take the Poisson rate λ to be R, the effective reproduction number, which is the expected number of cases directly caused by one case. In that case, the average number of cases following the Poisson distribution is
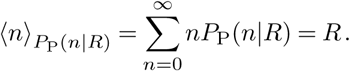 To account for variability in transmission dynamics, the basic Poisson distribution with a single rate R can be replaced with a continuous mixture of Poisson distributions, where the rate parameter λ follows a gamma distribution,
To account for variability in transmission dynamics, the basic Poisson distribution with a single rate R can be replaced with a continuous mixture of Poisson distributions, where the rate parameter λ follows a gamma distribution,
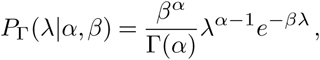 with shape parameter α and rate parameter β. The average value of λ is
with shape parameter α and rate parameter β. The average value of λ is
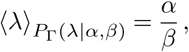 and its variance is
and its variance is
 In this context, it is natural to take α = k and β = k/R. With these choices, the gamma distribution reads
In this context, it is natural to take α = k and β = k/R. With these choices, the gamma distribution reads
 The parameter k is a dispersion parameter that determines how long-tailed the distribution is. The mean value of λ is always R, but when k is smaller its variance increases. In the limit that k → ∞, we recover the pure Poisson distribution with rate λ = R. When k = 1, the distribution of the number of cases n is geometric,
The parameter k is a dispersion parameter that determines how long-tailed the distribution is. The mean value of λ is always R, but when k is smaller its variance increases. In the limit that k → ∞, we recover the pure Poisson distribution with rate λ = R. When k = 1, the distribution of the number of cases n is geometric,
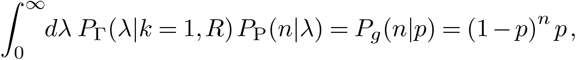 where p = 1/(1 + R). For arbitrary values of k > 0, the number of cases follows a negative binomial distribution,
where p = 1/(1 + R). For arbitrary values of k > 0, the number of cases follows a negative binomial distribution,
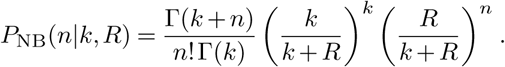 The standard parameters of the negative binomial distribution are r and p, which are set to k and k/(k + R) in our parameterization above.
The standard parameters of the negative binomial distribution are r and p, which are set to k and k/(k + R) in our parameterization above.
2.3. Dynamics for the pathogen variant frequencies
Let us assume that there exist multiple variants of a pathogen, which are distinguished by an index a. The number of cases infected with variant a is na. We assume that different variants have slightly different transmission probabilities, so that Ra = R(1 + wa), with |wa| ≪ 1. The term wa is analogous to a selection coefficient in population genetics.
2.3.1. Dynamics of multiple cases infected by a single variant
First, let us assume that n individuals, each labeled by an index i, are all infected by the same variant of a pathogen. For now we will assume that there is no travel into or out of the population, though we will include it later. How many cases will be generated from these individuals? The number of new cases for all individuals is
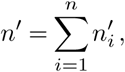 where the numbers of cases
where the numbers of cases 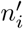 generated by individual i follows a negative binomial distribution. Because all individuals are infected by the same variant, the negative binomial parameter p = k/(k + R) is the same for each of them. Then, assuming that all of the infection events are independent, it can be shown that the probability distribution for the total number of new cases n′ also follows a negative binomial distribution with the same value of p, and with r = nk (that is, the new r parameter value is the sum of the individual r parameter values). Thus, the distribution of n′ is
generated by individual i follows a negative binomial distribution. Because all individuals are infected by the same variant, the negative binomial parameter p = k/(k + R) is the same for each of them. Then, assuming that all of the infection events are independent, it can be shown that the probability distribution for the total number of new cases n′ also follows a negative binomial distribution with the same value of p, and with r = nk (that is, the new r parameter value is the sum of the individual r parameter values). Thus, the distribution of n′ is
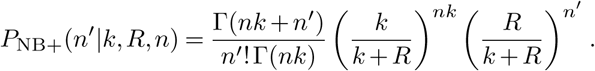
2.3.2. Dynamics for multiple cases infected by multiple variants
Let us extend the previous example to consider m variants of a pathogen. At the starting point, the number of individuals infected by a given variant a is na, with a ∈ {1,…, m}. The fraction of cases infected by variant a is
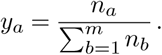 Now, we would like to know how the fraction of individuals infected by each variant is expected to change with each round of infections. In other words, for variant a, we would like to compute
Now, we would like to know how the fraction of individuals infected by each variant is expected to change with each round of infections. In other words, for variant a, we would like to compute
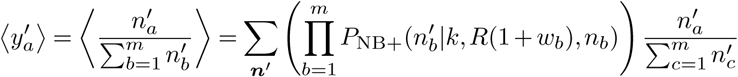 where the outer sum is over all vectors n′ with entries
where the outer sum is over all vectors n′ with entries 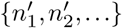 , and with
, and with  for all b. Here, we have assumed that the
for all b. Here, we have assumed that the  are independent across b.
are independent across b.
To proceed, it is convenient to write the negative binomial distributions as mixtures of Poisson distributions (as indicated above), giving
 Next, we use the fact that the sum of independent Poisson-distributed random variables is also Poisson with rate parameter equal to the sum of the individual rates, and that the distribution of independent Poisson random variables conditioned on their sum is multinomial, to write
Next, we use the fact that the sum of independent Poisson-distributed random variables is also Poisson with rate parameter equal to the sum of the individual rates, and that the distribution of independent Poisson random variables conditioned on their sum is multinomial, to write
 Here λ is a vector with entries λ1, λ2, …, and we have also introduced Σa λa = λ. Note also that the outer sum on the first line is over all vectors n′ whose (non-negative) entries sum to n′.
Here λ is a vector with entries λ1, λ2, …, and we have also introduced Σa λa = λ. Note also that the outer sum on the first line is over all vectors n′ whose (non-negative) entries sum to n′.
Computing the remaining integrals exactly is challenging, largely because the Gamma distributions have different rate parameters. To address this, next we will expand our expression to first order in the wa, since these are assumed to be small parameters. Referring back to Eq. (S1), the expansion gives
 Next we change variables to {λ, q1 = λ1/λ, q2 = λ2/λ,…, qm−1 = λm−1/λ}, because the distribution of the sum of gamma-distributed random variables, λ, with the same rate parameter and the ratios of the individual variables to the total (λa/λ) follow independent gamma and Dirichlet distributions3. The mth ratio
Next we change variables to {λ, q1 = λ1/λ, q2 = λ2/λ,…, qm−1 = λm−1/λ}, because the distribution of the sum of gamma-distributed random variables, λ, with the same rate parameter and the ratios of the individual variables to the total (λa/λ) follow independent gamma and Dirichlet distributions3. The mth ratio  by conservation. By convention we will also set wm = 0, which can be thought of as normalizing the value of R relative to a reference genotype. The transformation then gives
by conservation. By convention we will also set wm = 0, which can be thought of as normalizing the value of R relative to a reference genotype. The transformation then gives
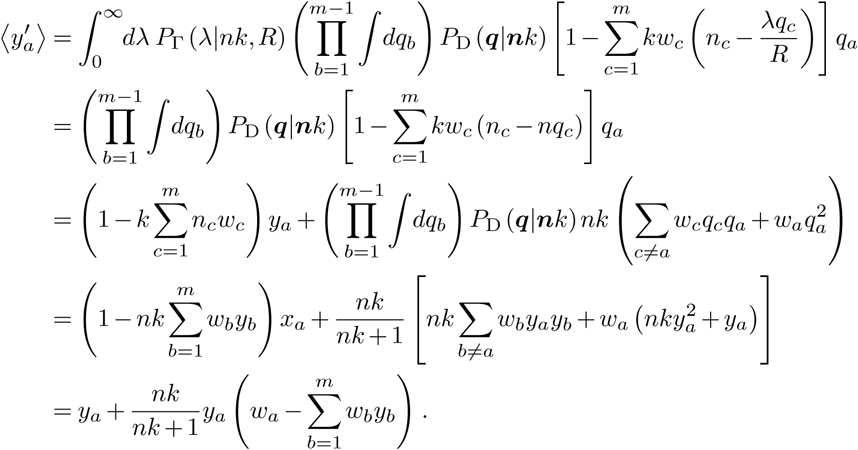 In the expressions above PD(q|α) is the Dirichlet distribution, with concentration parameters α given by nk in our case. Note that if wm ≠ 0, the last line should instead read
In the expressions above PD(q|α) is the Dirichlet distribution, with concentration parameters α given by nk in our case. Note that if wm ≠ 0, the last line should instead read
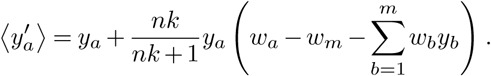 Thus, we obtain (with wm = 0)
Thus, we obtain (with wm = 0)
 Following a similar approach, we can compute the second moments. First, we consider
Following a similar approach, we can compute the second moments. First, we consider
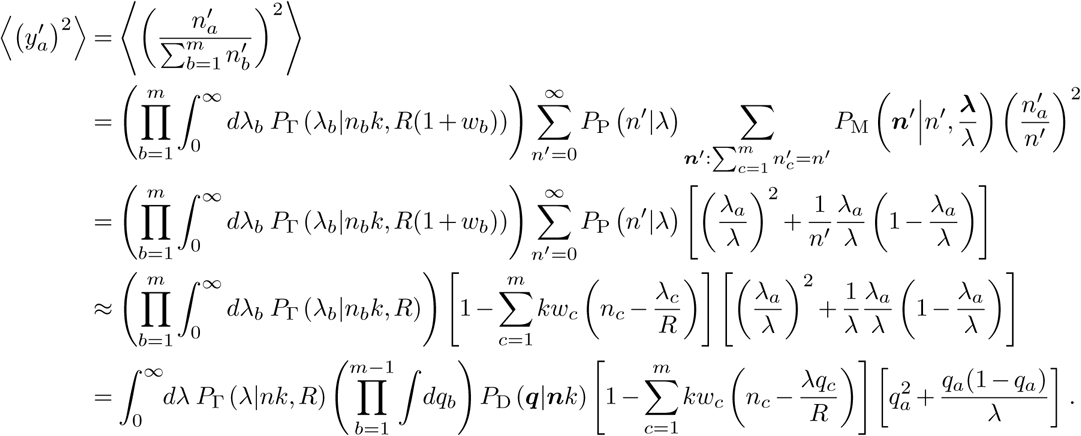 In going from the third to the fourth line above, we have made the approximation that
In going from the third to the fourth line above, we have made the approximation that
 which is valid for λ ≳ 1. Similarly,
which is valid for λ ≳ 1. Similarly,
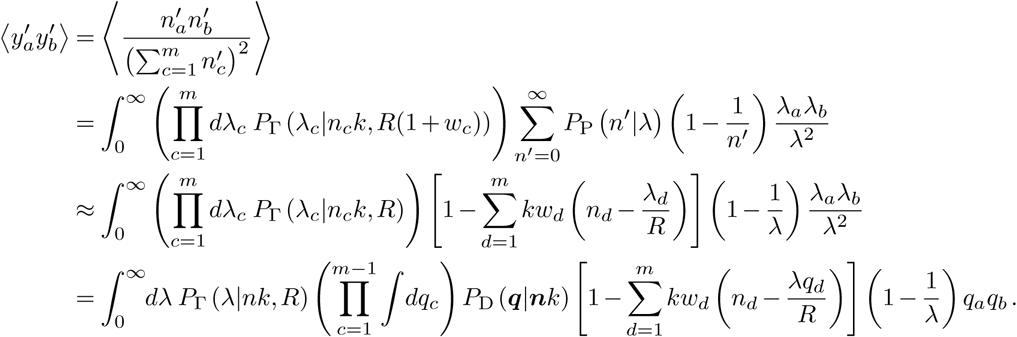 Simplifying the expressions above is tedious but straightforward. The following results are helpful:
Simplifying the expressions above is tedious but straightforward. The following results are helpful:
 Here we have frequently used na = nya to simplify expressions.
Here we have frequently used na = nya to simplify expressions.
With the above results, simplifying expressions for the second moments, we finally find
 and
and
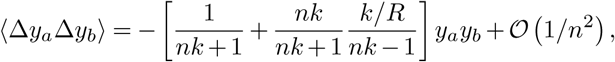 where we have assumed that the wa are 𝒪 (1/n), as in the Wright-Fisher model with weak selection. We have thus found that the first and second moments of frequency changes in our multi-variant epidemiological model have the same frequency dependence as those in the multispecies Wright-Fisher model, but with different scaling. The first moment (‘drift’) is multiplied by a factor of nk/(nk + 1), and the second moment (‘diffusion’) by
where we have assumed that the wa are 𝒪 (1/n), as in the Wright-Fisher model with weak selection. We have thus found that the first and second moments of frequency changes in our multi-variant epidemiological model have the same frequency dependence as those in the multispecies Wright-Fisher model, but with different scaling. The first moment (‘drift’) is multiplied by a factor of nk/(nk + 1), and the second moment (‘diffusion’) by
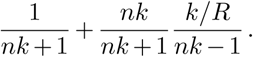 These prefactors match with the Wright-Fisher model exactly when k → ∞ (i.e., a pure Poisson distribution for the number of new cases per infected individual) and R = 1.
These prefactors match with the Wright-Fisher model exactly when k → ∞ (i.e., a pure Poisson distribution for the number of new cases per infected individual) and R = 1.
2.4. Correction to the first moment due to travel
Travel of infected individuals can change the total number of infected individuals in a region and will thus lead to a correction in the first and second moments that have been calculated above. Call na,in − na,out = δna. There will be a first order correction to the first moment due to fact that the number of individuals of variant a in the next generation is now 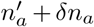 , where now
, where now  represents the number of individuals of variant a in the next generation that don’t come from travel. In addition, the total population in the next generation will be n′ + Σb δnb, where
represents the number of individuals of variant a in the next generation that don’t come from travel. In addition, the total population in the next generation will be n′ + Σb δnb, where 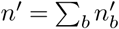 . Therefore, in calculating the first moment we will now have
. Therefore, in calculating the first moment we will now have
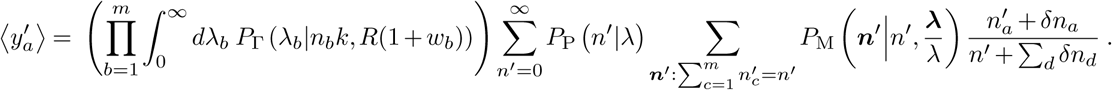 If we allow that the total inflow and outflow of a specific variant, δna, and of all variants together, Σd δnd, are both much smaller than the number of infected individuals, then the term on the far right can be expanded:
If we allow that the total inflow and outflow of a specific variant, δna, and of all variants together, Σd δnd, are both much smaller than the number of infected individuals, then the term on the far right can be expanded:
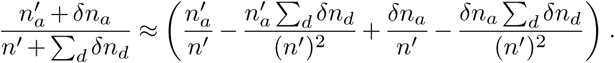 The first term simply reproduces the first order moment without travel. The last two terms lead to the correction
The first term simply reproduces the first order moment without travel. The last two terms lead to the correction
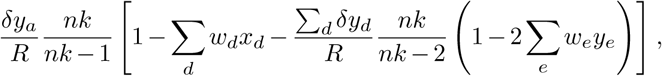 where δya = δna/n. Finally, for the second term, we have
where δya = δna/n. Finally, for the second term, we have
 Ultimately, this produces the additional correction
Ultimately, this produces the additional correction
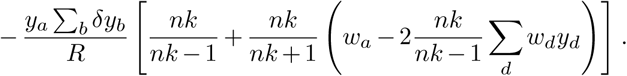 Remembering that both δyd and wd are small, the overall first order correction will be
Remembering that both δyd and wd are small, the overall first order correction will be

2.5. Correction to the second moment due to travel
There will also be a correction to the second moment due to travel, and for the diagonal terms, we will have,
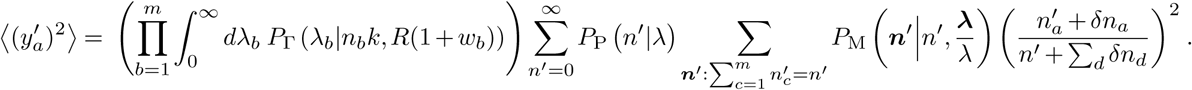 Again assuming that the number of infected individuals traveling is small compared to the total number of infected individuals in a region, we can expand the term on the far right:
Again assuming that the number of infected individuals traveling is small compared to the total number of infected individuals in a region, we can expand the term on the far right:
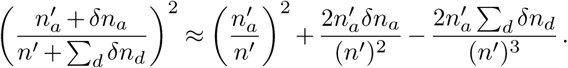 Similarly, we have
Similarly, we have
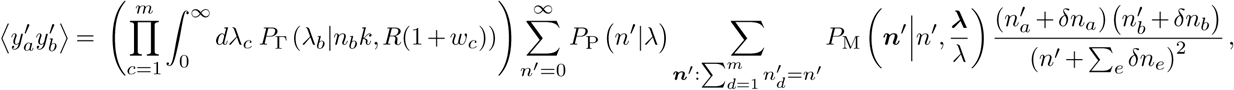 and the term on the far right can be expanded as
and the term on the far right can be expanded as
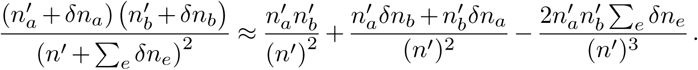 These can be calculated in much the same way as the correction to the first moment, and all of the resulting terms are 𝒪 (1/n2).
These can be calculated in much the same way as the correction to the first moment, and all of the resulting terms are 𝒪 (1/n2).
2.6. Derivation of the selection coefficient estimator
The derivation in this section closely follows that given in ref.4. It is well known that a WF process can be approximated by a continuous-time continuous-frequency diffusion process in the large n limit. In the continuous-time limit the time variable t has units of n generations, with one generation in discrete time taking τ = 1/N continuous time units. The change in the frequency of variant a in one generation due to travel is δya, so the change of frequency in τ generations is δya * τ. Furthermore, the selection coefficients wa are assumed to scale with n such that  , where
, where 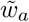 is a parameter independent of the population size n. In the limit of large population size, our generalized super-spreading model can, like the WF process, be approximated by a diffusion process, where the transition probability density ϕ is the solution to the Fokker-Planck equation
is a parameter independent of the population size n. In the limit of large population size, our generalized super-spreading model can, like the WF process, be approximated by a diffusion process, where the transition probability density ϕ is the solution to the Fokker-Planck equation
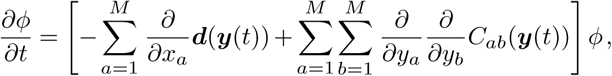 where M is the number of distinct genotypes, y is the genotype frequency vector, d is the drift vector, and C is the diffusion matrix. Ignoring recombination and mutation, since these are comparatively small and therefore unlikely to significantly affect estimates of changes in viral transmission (though these can be included and the solution remains tractable), the drift and diffusion have entries given by,
where M is the number of distinct genotypes, y is the genotype frequency vector, d is the drift vector, and C is the diffusion matrix. Ignoring recombination and mutation, since these are comparatively small and therefore unlikely to significantly affect estimates of changes in viral transmission (though these can be included and the solution remains tractable), the drift and diffusion have entries given by,
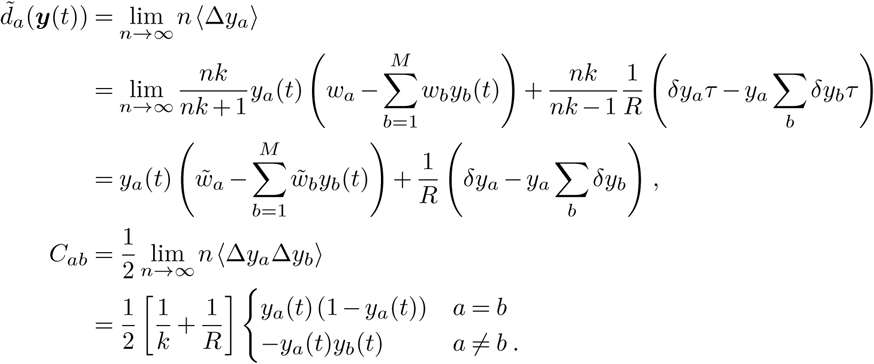 The Fokker-Planck equation can be converted into a path integral approximation for the transition probability density
The Fokker-Planck equation can be converted into a path integral approximation for the transition probability density
 We write the re-scaled drift vector as
We write the re-scaled drift vector as 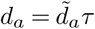 . Since we aim to infer selection coefficients for the SNVs, it is more convenient to work with the allele frequencies xi instead of the genotype frequencies ya. The allele frequency at site i is given by
. Since we aim to infer selection coefficients for the SNVs, it is more convenient to work with the allele frequencies xi instead of the genotype frequencies ya. The allele frequency at site i is given by
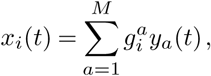 where
where 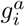 is a 1 if there there is a mutant allele at site i on genome a and zero if there is not. Similarly, if the selection coefficient for the genotype a is wa and the allele level selection coefficient for allele i is si, then they are related by:
is a 1 if there there is a mutant allele at site i on genome a and zero if there is not. Similarly, if the selection coefficient for the genotype a is wa and the allele level selection coefficient for allele i is si, then they are related by:
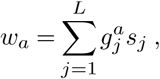 where L is the length of the genome.
where L is the length of the genome.
The allele level drift and diffusion terms will be linear combinations of the genotype level drift and diffusion, just as with the frequencies and the selection coefficients. The drift vector for the allele frequencies can be transformed by
 The sum in the last term can be interpreted as the total number of infected individuals added or subtracted to the population due to travel, divided by the population size. This can be used, along with the transition probability density for genomes, in order to find an approximation for the mutant allele transition probability density:
The sum in the last term can be interpreted as the total number of infected individuals added or subtracted to the population due to travel, divided by the population size. This can be used, along with the transition probability density for genomes, in order to find an approximation for the mutant allele transition probability density:
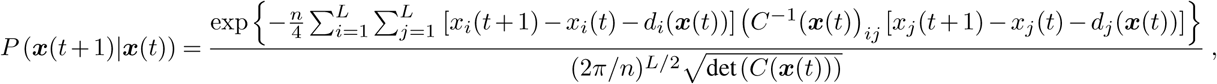 where here the diffusion C is derived similarly to the drift d and has entries
where here the diffusion C is derived similarly to the drift d and has entries
 A path integral then gives the probability of observing a trajectory of allele frequencies (x(t1), x(t2), …, x(tK)), and is given by
A path integral then gives the probability of observing a trajectory of allele frequencies (x(t1), x(t2), …, x(tK)), and is given by
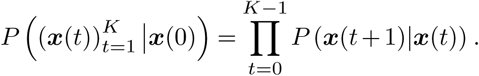 Bayesian analysis can then be used to show that the posterior probability of the selection coefficients s = (s1, s2, …, sL) given an observed frequency path x(0), x(1), …, x(T) is
Bayesian analysis can then be used to show that the posterior probability of the selection coefficients s = (s1, s2, …, sL) given an observed frequency path x(0), x(1), …, x(T) is
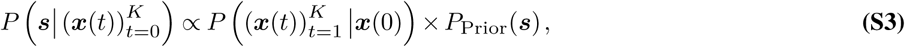 where we use a Gaussian prior distribution with zero mean and adjustable covariance determined by the parameter γ.
where we use a Gaussian prior distribution with zero mean and adjustable covariance determined by the parameter γ.
For the inferred coefficients, we take those that maximize the posterior probability. They can be analytically found by a simple application of the Euler-Lagrange equations to equation S3 and are given by
 The second term in the numerator is the correction due to travel of infected individuals into and out of the region, and is given by
The second term in the numerator is the correction due to travel of infected individuals into and out of the region, and is given by
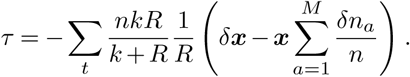
2.7. Extension to multiple regions
In the SARS-CoV-2 pandemic, and in real disease outbreaks in general, there are frequently multiple different outbreaks in different regions that develop largely or entirely independently of one another. In order to find the best estimate for the selection coefficients using the data from multiple regions, the estimator can be generalized to find the maximum a posteriori estimate for the selection coefficients given the time series of allele frequencies in each of the regions. If the probability for a specific path in a specific region r is given by 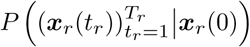 , where xr is the allele frequency vector in region r, then the joint probability of the specific paths in all of the regions is simply the product of the individual region probabilities:
, where xr is the allele frequency vector in region r, then the joint probability of the specific paths in all of the regions is simply the product of the individual region probabilities:
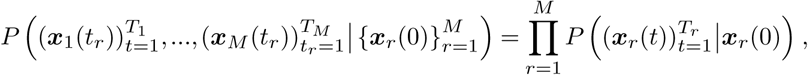 where M is the number of different regions. Since this is a product of exponential functions, the log posterior will be the sum of the exponents and the regularization. This can be maximized with respect to the selection coefficient vector s as before, which, dropping the travel term, leads to the estimator:
where M is the number of different regions. Since this is a product of exponential functions, the log posterior will be the sum of the exponents and the regularization. This can be maximized with respect to the selection coefficient vector s as before, which, dropping the travel term, leads to the estimator:

2.8. Simplification of the estimator
In real outbreaks the parameters k, R, and n are in general time-varying. In our simulations as well, R and n are time-varying (and k can be constant or time-varying). In order to accurately infer the selection coefficients according to Eq. (S4) or Eq. (S5), it would seem that we need to accurately infer the values of k, R, and N at every point in the time series. In practice, this would be extremely difficult. For general discussion about the effective reproduction number R and the basic reproduction number Rt as well as some attempts to infer this, see refs.5–9. In order to get an accurate estimate for k it is necessary to have pervasive contact tracing, so that the negative binomial distribution is well sampled, and there are other difficulties in inferring k as well10–12. Lastly, it can be difficult to estimate the number of new infections due to multiple factors, including the difference between the population that gets tested and the population that does not, test result inaccuracies, and delays between symptom onset, testing, and reporting13,14.
We propose an alternative that lets us avoid these complications. The prefactor nkR/(R + k), multiplies both the numerator and the denominator. Therefore, the only effect of the prefactor is to weight time points more heavily if the population size, the dispersion parameter, or the basic reproduction number, is larger. This makes sense in theory, because a larger n or k implies that there is less noise and the trajectories are more deterministic, while a larger R means that there are more new infections per generation and thus more data to use to infer the selection coefficients. This does hold with perfect information, that is, if all infected individuals are sampled at every time point. However, in practice, finite sampling is the source of significantly more noise than that due to a time-varying population size or dispersion, so weighting the time points based upon n, k, or R in fact leads to worse inference than assuming the parameters are constant in time and thus weighting the time points equally. However, in the special and unrealistic case of perfect sampling, using the actual parameters does lead to better inference than using constant parameters (see Supplementary Fig. 4). If the time points are weighted equally, then, provided that the regularization γ is scaled appropriately (and in general it must be determined by separate means, discussed below), the prefactors in the numerator and denominator cancel, and the estimator is independent of n, k, and R. Defining  by
by
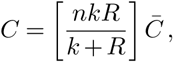 so that
so that
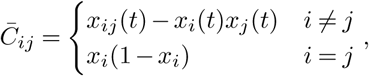 Eqs. (S4) and (S5) for the selection coefficients become, respectively
Eqs. (S4) and (S5) for the selection coefficients become, respectively
 which are the same as the MPL estimators for the Wright-Fisher model except that we have ignored the mutation term because the mutation rate for SARS-CoV-2 is small4.
which are the same as the MPL estimators for the Wright-Fisher model except that we have ignored the mutation term because the mutation rate for SARS-CoV-2 is small4.
3. Simulations
We tested the inference using simulations of disease spread. We used two different kinds of simulations. The first is the super-spreader simulation based on the model described above, which is an analog of the Wright-Fisher model where the sampling distribution for the number of new infections per infected individual is drawn from a negative binomial distribution instead of a pure Poisson distribution The second is a standard deterministic susceptible-infected-recovered (SIR) model that has been adapted in order to include multiple variants of the virus with different transmission rates, described in Methods.
3.1. Description of simulations
We simulated disease spread as a branching process in which the number of individuals infected per currently infected individual is drawn from a negative binomial distribution whose shape is determined by the basic reproduction number R0 (or the reproduction number, R, in a population that is not totally susceptible) and the dispersion parameter k. Because we sample in this way, the population size is not constant. However, if the population size is too small, then the population is extremely likely to die off stochastically, and if the population size is too large, then sampling from the negative binomial becomes too computationally expensive. In order to avoid both of these problems, once the population size is large enough R is adaptively adjusted so that the average reproduction number for the entire population will remain near 1, and the population size will oscillate around a fixed value. An explicit time-varying population size can also be used as input, and R will be adaptively adjusted to remain near the given curve. Constant values can be used for the dispersion k or k can vary as a function of time, perhaps representing different degrees of social distancing or lockdown measures at different times. Since different interventions implemented to prevent the spread of disease would likely affect the shape of the distribution of the number of individuals infected by a single infected individual, time-varying values for k and R can be used to reflect these effects. We also implement travel of specified variants into or out of the population over time.
3.2. Inference
The simulations are run for a number of generations and genomes are sampled from the population of infected individuals at different times using a multinomial sampling distribution. This sampled time series is then used to infer the selection coefficients using Eq. (S4). Alternatively, multiple simulations can be run and the joint inference of the selection coefficients can be made using Eq. (S5). We find that, given good enough sampling, a long enough time series, and sampling that occurs at a sufficient number of times, the selection coefficients can be inferred very accurately (Fig. 1). The quality of inference is significantly improved if multiple simulations are combined and if mutated sites show up in more than one of the simulations, even under less than ideal sampling conditions. Beneficial coefficients are typically inferred more accurately than deleterious ones, likely because deleterious SNVs frequently die off and therefore there is less data to use for inference.
The inference is robust to shortening the time-series or lowering the number of samples taken per generation, though obviously if either of these conditions is too extreme (or worse, both), the inference starts to break down. The negative effects of a short time-series or poor sampling can be somewhat made up for by using multiple simulations, which is analogous to using data from outbreaks in multiple regions. In addition, the diffusion approximation is only valid in the large n limit. However, we tested the inference for small population sizes and found that inference is accurate even if the population of newly infected individuals per serial interval is as low as a few hundred (Fig. 1).
It is reasonable to expect that in a real outbreak there will be some travel of infected individuals into and out of the population. This can affect the estimation of the selection coefficients if the travel term, Eq. (S2), is ignored. As a simplified example, imagine there is a steady influx of a variant that has a SNV that is entirely neutral, and little to no outflow of this variant. In this case the selection coefficient for the SNV that migrates into the population will likely be overestimated because the frequency of the SNV will in general be increasing, even though this increase in frequency is not due to a selective advantage. Similarly, if there is an excess of outflow of a certain neutral SNV, then the fitness for this SNV will in general be underestimated. Testing this with simulations, we found that modest influx of a neutral SNV over a long time (25 importations per serial interval compared to n = 104 local transmissions, continuing for 100 serial intervals) produces a small but detectable bias in the inferred selection coefficient, which can be corrected by including the travel term and using the true flux of variants for δna (Supplementary Fig. 8). More generally, corrections due to travel should become significant when the term τint becomes large compared to observed changes in SNV frequencies.
ACKNOWLEDGEMENTS
We gratefully acknowledge the numerous laboratories worldwide that have provided sequence data and metadata to GISAID. A full list of originating and submitting laboratories for the sequences used in our analysis can be found at https://www.gisaid.org using the EPI-SET-ID: EPI_SET_20211201ze. The work of B.L., E.F., and J.P.B. reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R35GM138233. The work of S.F.A., M.S.S., A.A.Q., and M.R.M. was supported by the General Research Fund of the Hong Kong Research Grants Council under Grant Number 16213121.
References
References



