
Summary
In a study of 207 SARS-CoV2-infected individuals with a range of severities followed over 12 weeks from symptom onset, we demonstrate that an early robust immune response, without systemic inflammation, is characteristic of asymptomatic or mild disease. Those presenting to hospital had delayed adaptive responses and systemic inflammation already evident at around symptom onset. Such early evidence of inflammation suggests immunopathology may be inevitable in some individuals, or that preventative intervention might be needed before symptom onset. Viral load does not correlate with the development of this pathological response, but does with its subsequent severity. Immune recovery is complex, with profound persistent cellular abnormalities correlating with a change in the nature of the inflammatory response, where signatures characteristic of increased oxidative phosphorylation and reactive-oxygen species-associated inflammation replace those driven by TNF and IL-6. These late immunometabolic inflammatory changes and unresolved immune cell defects, if persistent, may contribute to “long COVID”.
Introduction
The immune pathology associated with COVID-19 is complex (Wang et al., 2020; Zhou et al., 2020). Most infected individuals mount a successful anti-viral response, resulting in few if any symptoms. In a minority of patients there is evidence that ongoing cytokine production develops, associated with persistent systemic inflammation, end-organ damage and often death (Del Valle et al., 2020; Lucas et al., 2020). The relationship between the initial immune response to SARS-CoV-2, viral clearance, and development of the ongoing inflammatory disease which drives severe COVID-19 is not clearly established, nor have the kinetics of the immune changes seen in COVID-19 been fully assessed as disease progresses. Defective immune recovery might drive ongoing disease, and contribute to long-term disease sequelae (“long COVID”) and perhaps to secondary immunodeficiency with an increased risk of subsequent infection.
Severe COVID-19 is associated with profound abnormalities in circulating immune cell subsets (Arunachalam et al., 2020; Hadjadj et al., 2020; Kuri-Cervantes et al., 2020; Laing et al., 2020; Mann et al., 2020; Mathew et al., 2020; Maucourant et al., 2020; Schulte-Schrepping et al., 2020; Su et al., 2020; Wen et al., 2020). There is a decrease in many peripheral blood subsets of both CD4 and CD8 T cells (Kuri-Cervantes et al., 2020; Laing et al., 2020; Mann et al., 2020; Mathew et al., 2020; Su et al., 2020), but an increase in activated and differentiated effector cells (Arunachalam et al., 2020; Hadjadj et al., 2020; Kuri-Cervantes et al., 2020; Laing et al., 2020; Mann et al., 2020; Mathew et al., 2020; Su et al., 2020). Cells expressing PD1 and other inhibitory molecules are increased, though whether these reflect genuine T cell exhaustion or changes accompanying T cell activation, has not been firmly established (Hadjadj et al., 2020; Laing et al., 2020; Mathew et al., 2020; Su et al., 2020; Zheng et al., 2020). There is, nonetheless, evidence of functional impairment in both CD8 and CD4 T cells in a number of studies (Chen and Wherry, 2020). Data on T helper cell subsets is variable, but there is evidence of increased TH17 cells and markedly reduced T follicular helper cells (TFH) (Kuri-Cervantes et al., 2020; Mathew et al., 2020; Su et al., 2020). There have been conflicting reports regarding B cell immunity (Laing et al., 2020; Mann et al., 2020; Mathew et al., 2020), but increased circulating plasmablasts (Arunachalam et al., 2020; Hadjadj et al., 2020; Laing et al., 2020; Mathew et al., 2020) and reduced germinal centre responses (Su et al., 2020) are consistently observed in severe COVID-19. A number of innate T cell subsets, including γd T cells and MAIT cells, are also reduced (Kuri-Cervantes et al., 2020; Laing et al., 2020; Maucourant et al., 2020; Parrot et al., 2020), as are non-classical monocytes (Schulte-Schrepping et al., 2020; Su et al., 2020) and both plasmacytoid and myeloid dendritic cells (Kuri-Cervantes et al., 2020; Laing et al., 2020).
By analysing longitudinal samples from COVID-19 patients with a range of disease severities, for up to 3 months from symptom onset, we were able to address two important questions regarding the immune response to SARS-CoV-2: (i) How does the very early immune response in patients who cleared virus and recovered from disease with few or no symptoms, compare with those who progressed to severe inflammatory disease. This provided insight into what constitutes an effective versus an ineffective immune response, and whether systemic inflammation is an early or later development in those who progress to severe disease. (ii) How rapidly do the profound immune defects that accompany severe COVID-19 recover, and do the kinetics of recovery relate to ongoing inflammation and clinical status.
We recruited 207 patients with COVID-19, ranging from asymptomatic healthcare workers in whom SARS-CoV-2 was detected on routine screening, through to patients requiring assisted ventilation, and compared their results to 45 healthy controls. We performed detailed immune phenotyping at multiple time points up to 90 days from symptom onset, reporting absolute cell counts rather than proportions as the latter are difficult to interpret in many studies in the context of profound lymphopenia. Correlation of these data with clinical and other meta-data demonstrated that the immune response in patients with progressive COVID-19 is delayed compared to those with mild disease, and is inflammatory in nature from the outset. Early immune cellular changes predict severe disease course. Their variable recovery over 3 months is associated with marked changes in the nature of the systemic inflammation seen in severe COVID-19.
Results
Patient cohorts
SARS-CoV-2 PCR positive subjects were recruited for this study between 31st March and 20th July 2020 in three contexts. Those without symptoms, or with mild symptoms, were recruited from routine screening of healthcare workers (HCW) at Addenbrooke’s Hospital (Rivett et al., 2020). COVID-19 patients were recruited at presentation to Addenbrooke’s hospital if their symptoms were consistent with COVID-19, and then formally included for follow-up if subsequent swab results were positive for SARS-CoV-2. In addition, some patients were recruited having already been admitted, twenty-nine having developed COVID-19 in hospital after admission for another reason, with others recruited after transfer to intensive care at either Addenbrooke’s or Royal Papworth hospitals. After recruitment patients were bled approximately weekly, and then invited to an outpatient follow-up visit 4-12 weeks after study enrolment. HCWs were sampled at study entry, and then approximately 2 and 4 weeks later.
Study participants were divided into five categories according to clinical severity, which we use throughout this paper unless otherwise stated (Figures 1A and S1). These were:
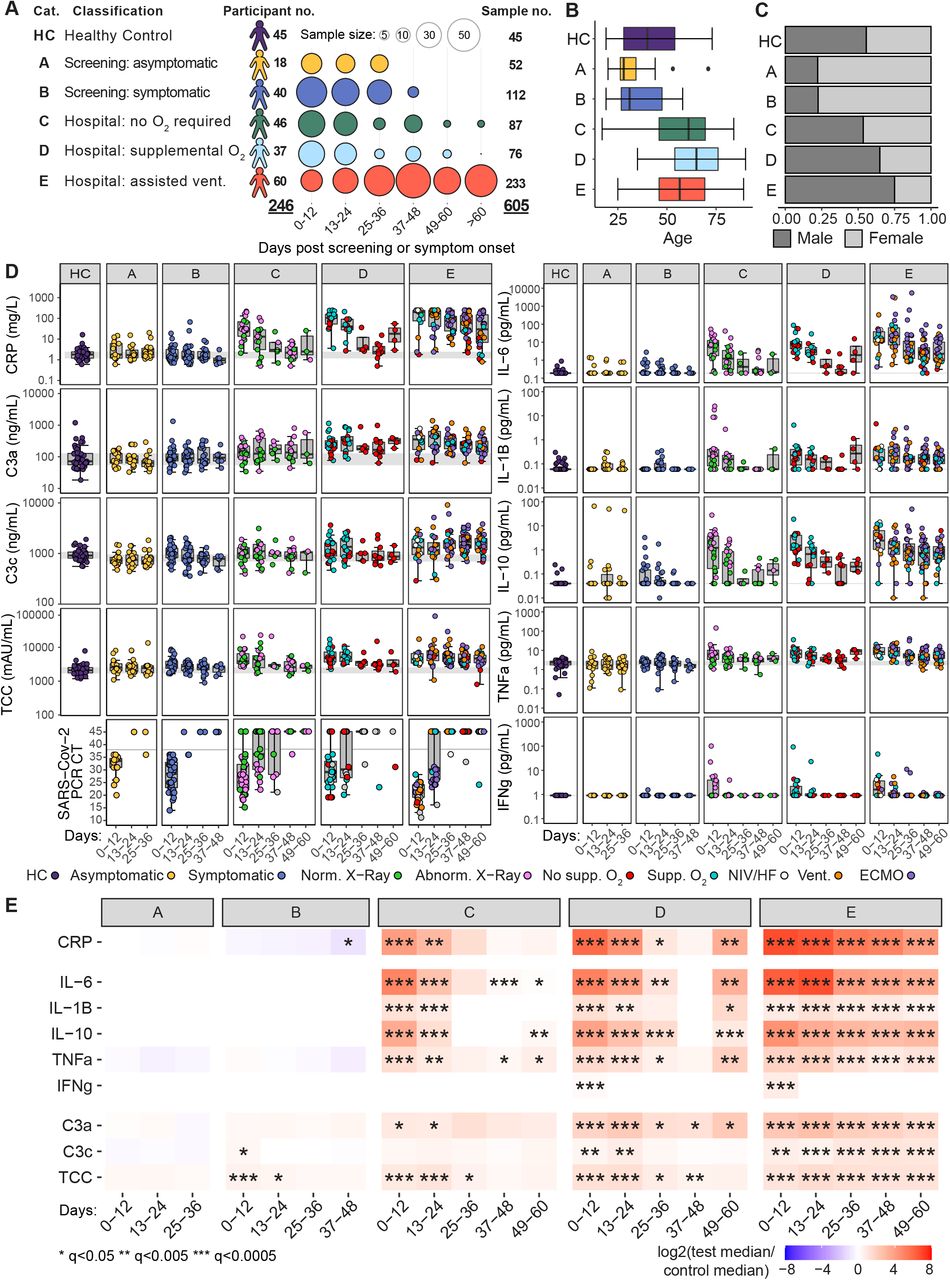
A) Study participant and sample numbers split by severity categories and 12-day time bins post screening (group A) or symptom onset (group B-E). Distribution of participant age B) and gender C) across severity categories. D) Boxplots showing measured CRP (mg/L), complement proteins, cytokines and SARS-CoV-2 PCR cycle threshold (CT) for samples collected within 12-day time bins. Grey band indicates the interquartile range of the corresponding measure in HCs, or the SARS-CoV2 negative swab cycle threshold (CT > 38). Points are coloured based on asymptomatic or symptomatic classification for categories A and B respectively, normal or abnormal chest radiology (group C), and mode of respiratory support at sampling (group D and E); time points missing respiratory status are coloured grey. E) Heatmap showing log2 fold change in median measure between COVID-19 cases and HC, within severity categories and across 12-day time bins. Wilcoxon test FDR adjusted p-value: *<0.05, **<0.005, ***<0.0005. CT, cycle threshold; CRP, C-reactive protein; IL, interleukin; TNF, tumour necrosis factor; IFN, interferon; C3, complement 3; TCC, terminal complement complex; NIV/HF, non-invasive ventilation/high-flow oxygen; ECMO, extracorporeal membrane oxygenation.
A) asymptomatic HCWs.
B) HCWs who either were still working with mild symptoms insufficient to meet the criteria for self-isolation (Rivett et al., 2020), or who were symptomatic and self-isolating.
C) patients who presented to hospital but never required oxygen supplementation.
D) patients who were admitted to hospital and whose maximal respiratory support was supplemental oxygen.
E) patients who at some point required assisted ventilation. Three patients who died without admission to intensive care were also included in this severe group.
In addition, 45 healthy controls were recruited, distributed across a range of age and gender.
This study analysed the immune phenotype of 605 blood samples from 246 participants out to 90 days from the onset of symptoms (Figure 1A). Patients were included irrespective of co-morbidity to reflect “real world” disease (Table S1), apart from the exclusion of 6 patients whose severe non-COVID-19 disease determined their clinical outcome, which made investigation results uninterpretable, and in whom COVID-19 was essentially a side-issue (details in Table S2). It is important to note that as the clinical severity category increased, patients were more likely to be older and to be male (Figures 1B and 1C), as expected (Wang et al., 2020). A high-sensitivity C-reactive protein (CRP) assay was performed on all samples, and is shown for each cohort - demonstrating that classifying disease severity by subdivision on the basis of maximal respiratory support is reflected in the CRP (Figure 1D). Time is measured since the first positive swab for cohort A, and since the onset of symptoms for other cohorts, and CRP (and, later, other variables such as cell number) is compared to the interquartile range of 45 healthy controls. Nasopharyngeal swabs were assessed for SARS-CoV-2, allowing diagnosis and inclusion in this study, and were repeated in some patients at various times. Initial viral titres (higher in those with low PCR cycle threshold or CT values) were higher in group E. With only occasional exceptions, patients in all severity groups had cleared virus by 24 days after the onset of symptoms (Figures 1D and S1D). Of the 6 patients with positive swabs after 30 days, four were overtly immunosuppressed (3 solid organ transplants with recent induction/rejection treatment, 1 myeloma on B-cell depletion therapy) and one was a peritoneal dialysis patient admitted with peritonitis.
We will first outline the major datasets collected in this study, before integrating them to study early and recovering disease.
Cytokines and complement components
We assessed cytokine and complement components in plasma at each time point (Figures 1D and 1E). Asymptomatic HCWs in group A had no evidence of cytokine or complement dysregulation, while those with mild symptoms (group B) showed an early, transient increase in C3c and the terminal complement complex (TCC), but not in C-reactive protein (CRP) or cytokine levels. Once patients developed symptoms severe enough to warrant attendance at hospital (group C or above), a different picture was apparent. Both IL-6 and TNF-α were significantly raised, along with other cytokines, as were all of the complement components measured. These abnormalities were maximal at the first bleed, and largely persisted in group E, where many patients remained in intensive care throughout their course. Abnormalities in both IL-6 and TNF-α persisted in groups C and D despite clinical improvement (all had been discharged by the 49-60 time window). Interferon-gamma (IFN-γ) was raised in only a subset of patients, and in all severity groups this increase was short-lived.
Increased C3c was prominent in those with mild symptoms (group B), while C3a became the dominant complement component elevated in more severe disease (groups C –E). Cytokine levels vary with disease severity and over time, with IL-6, TNF-α, IL-10 and IL-1β rising in those with more severe disease (groups C-E). In contrast, there is no evidence of increased inflammatory cytokines in patients from groups A and B, pointing to marked differences in very early immune responses between resolving and progressive disease. In addition, the persistence of cytokine abnormalities even beyond 60 days from symptom onset could have implications for resolution of both immune abnormalities and clinical disease.
Both onset and recovery of immune cellular abnormalities vary with disease severity
Using standardised flow cytometry panels, we explored the size of 65 cell populations over time across the five clinical strata. Trucount analysis enabled calculation of absolute cell numbers. Cellular changes were assessed across time “bins” of 12 days (using the earliest measure per patient per bin in instances of repeat sampling), with four cell subsets shown in Figure 2A as examples. The outcomes for 30 cell types are summarised in a heat map, showing changes in cell population size relative to the median for healthy controls (Figure 2B). CyTOF, which uses whole blood rather than peripheral blood mononuclear cells (PBMCs), was also used in a subset of patients, as this allows quantification of granulocytes (largely absent in PBMCs) and non-classical and intermediate monocytes (both variably lost in PBMC separation: Figure 2B and Methods). Apart from these exceptions, cell numbers generated by CyTOF correlated well with data from flow cytometry (Figure S2). Data for all cell types, also including time as a continuous variable, is shown in Figure S3. Also shown in Figure S3 are samples taken beyond 48 days which, with the exception of group E, were not numerous enough for statistically useful inclusion in the “binned” data.

A) Boxplots showing absolute counts (cells/uL) for four representative cell populations, split by severity categories and 12-day time bins post screening (group A) or symptom onset (group B-E). Grey band indicates the interquartile range of the corresponding population in HC. Points are coloured based on asymptomatic or symptomatic classification for categories A and B respectively, normal or abnormal chest radiology (group C), and type of respiratory support at time of sampling (group D and E). B) Heatmap showing the log2 fold change in median absolute cell count between COVID-19 cases and HCs, within severity categories and across 12-day time bins. Wilcoxon test FDR adjusted p-value: *<0.05, **<0.005, ***<0.0005. Population hierarchy and associated cell surface markers are shown to the left. PBMC, peripheral blood mononuclear cells, analysed by flow cytometry; WB, whole blood, analysed by CyTOF.
Few changes in the immune phenotypes were seen in patients with asymptomatic (A) and very mild (B) disease, but once symptoms had become sufficient to warrant presentation to hospital, the picture changed markedly (see below). There were widespread immune abnormalities in patients with moderate through to severe disease (C-E), most marked at the time of first blood sampling (Figure 2B), even when this coincided with or was within a day or two of symptom onset (Figure S3). Almost all CD4 T cells subsets were reduced, as were many CD8 T cell subsets and both naive and memory B cells. A number of innate lymphoid subsets were also reduced, including MAIT cells, various γd T cell subsets, and NK cells. The myeloid compartment was also affected, with a reduction in myeloid dendritic cells, and both non-classical and intermediate monocytes. These changes were correlated with, and were predictive of, severity, as discussed below.
Blood transcriptomic inflammation-related signatures vary with severity and time
RNA was prepared from whole blood collected in PAXGene tubes at each bleed. RNA was isolated, and whole blood transcriptomes were generated by RNA-Sequencing (see Methods) and analysed in two time “bins” – 0 to 24 days and 25 to 48 days (finer gradations were not possible due to sample size). We first analysed the transcriptome data using PLIER which performs matrix factorization to identify interpretable latent factors. The contribution to each latent factor by immune cell subsets was then calculated across the severity groups and time points (Figures 3A and S4A). These RNA-Seq-derived latent factors were broadly aligned with the pattern observed in the cell count data (Figure 2B). An exception to this was the pronounced neutrophil signature seen at day 0 to 24 across groups C to E, and persisting at day 25-48 in group E. This transcriptomic analysis shows more pronounced neutrophil dysregulation across severity categories than is suggested by increasing neutrophil number alone. An erythrocyte gene expression-driven latent factor was also seen, and was prominent in group E at late times. This may be associated with heme metabolism, and is discussed below.

A) Cell subset deconvolution performed using PLIER, leveraging off prior knowledge of cell specific pathways. COVID-19 cases split by severity categories and 24-day time bins. Latent factor expression compared with HC, FDR adjusted p-value: *<0.05, **<0.005, ***<0.0005. B) Heatmap derived from WGCNA, illustrating the correlation of whole blood co-expression gene modules (coloured blocks, y axis) with COVID-19 severity groups (x axis) split by 24-day time bins. Boxplots displaying eigengene of key transcriptomic modules according to disease severity and time. Boxes are coloured by strength of correlation. C) Mixed-effects model with quadratic time trend showing the longitudinal expression of key eigengene over time, grouped by severity. Grey band indicates the interquartile range of the corresponding eigengene in HCs. Nominal and adjusted p-values for the time x severity group interaction term are reported. D) Equivalent mixed-effects model showing changes in SARS-CoV-2 PCR cycle threshold (viral load), over time across severity groups. Y-axis is inversed. E) GSEA assessing enrichment for HALLMARK genesets against HC in COVID-19 cases split by severity categories and 24-day time bins, FDR adjusted p-value: *<0.2, **<0.1, ***<0.05, ****<0.01. CT, cycle threshold; ISG, interferon stimulated genes.
We then used weighted gene correlation network analysis (WGCNA) to identify, in an unbiased fashion, modules of co-regulated genes in the whole blood transcriptome data, where each module can be summarised as an “eigengene”. Prominent gene expression modules were observed, that correlated with both disease severity and time (Figures 3B, S4B, S4C and Table S3). It can be seen that the module enriched for TNF-α /IL-6 genes correlates well with the cytokine levels determined in Figure 1 – rising early in groups C-E and then largely resolving by 25-48 days. A neutrophil activation module was also prominent early across groups C to E, as was one associated with glycolysis. Thus, there is clear transcriptional evidence of activation of broad inflammatory pathways at early time points, and these largely recover in most patient groups (with the exception of group E, in which many patients have persistent disease). In contrast, an interferon-related module is upregulated prominently in groups B-E at day 0 to 24 from symptom onset, but declines at later time points. As previously described (Banchereau et al., 2017), the relative contributions to this module by Type I, II and III interferons cannot be easily distinguished at the transcriptome level. A more detailed analysis of the kinetics of this interferon-stimulated gene (ISG) – associated module shows that, while expression peaks at different levels in each severity group it then declines in all of them by around 30 days (Figure 3C), coincident with viral clearance and occurring irrespective of clinical and inflammatory state (Figure 3D).
Finally, a supervised gene set enrichment analysis (GSEA) was performed using publicly available Hallmark gene signatures (Figure 3E) (Liberzon et al., 2015). These findings were largely consistent with those generated from the unbiased approaches above, and demonstrated a late upregulation of genes associated with reactive oxygen species and oxidative phosphorylation. Late upregulation of these pathways is discussed below in the context of immune recovery.
Immune phenotype at presentation correlates with severity and may predict outcome.
To determine if the immune phenotype at presentation correlated with, or indeed could predict, subsequent disease course, we first performed a Principal Component Analysis using cell numbers across 24 primary immune cell populations from blood draws taken between 0 and 10 days after the development of symptoms. Study participants in groups A and B clustered together with HC and were separate from those in groups C-E (Figure 4A). Hierarchical clustering of absolute cell counts from COVID-19 cases identified two clear clusters (Figure 4B), one almost entirely comprised of HCWs from groups A and B (cluster 2), and the other containing all patients who progressed to ventilation and/or death, and most who required supplementary oxygen support (cluster 1). The severe cluster 1 was associated with increased age, CRP, TNF-α and IL-6 (Figures 4C and S5A). Early differences between cell types drive this clustering, despite different cell subsets having quite different subsequent trajectories (Figure S5B). To determine which specific immune subsets drove this clustering, we used a sparse partial least squares discriminant analysis (sPLS-DA), which showed patient clusters 1 and 2 could be discriminated with a minimum classification error rate of 0.07 ± 0.02 (93% accuracy) based on 13 key cell populations selected by the model as most informative for cluster prediction (Figures 4D, S5C-E). The area under the receiver operator characteristic curve (AUROC) for patient cluster classification based on these 13 cell types was 0.98 (98% chance of accurate cluster prediction) (Figure 4E). These cell types were often the most profoundly affected by severe COVID-19, and those most associated tended to recover poorly over time (particularly MAIT, γδ T cells, TFH and CD4 EMRA T cells) suggesting a persistent association with disease severity. We also clustered patients using RNA-Seq data obtained from 1-10 days after onset – this largely recapitulated that seen using immune cell number, and was driven by ISG, TNF-α and IL-6 associated gene pathways (Figure 4F).
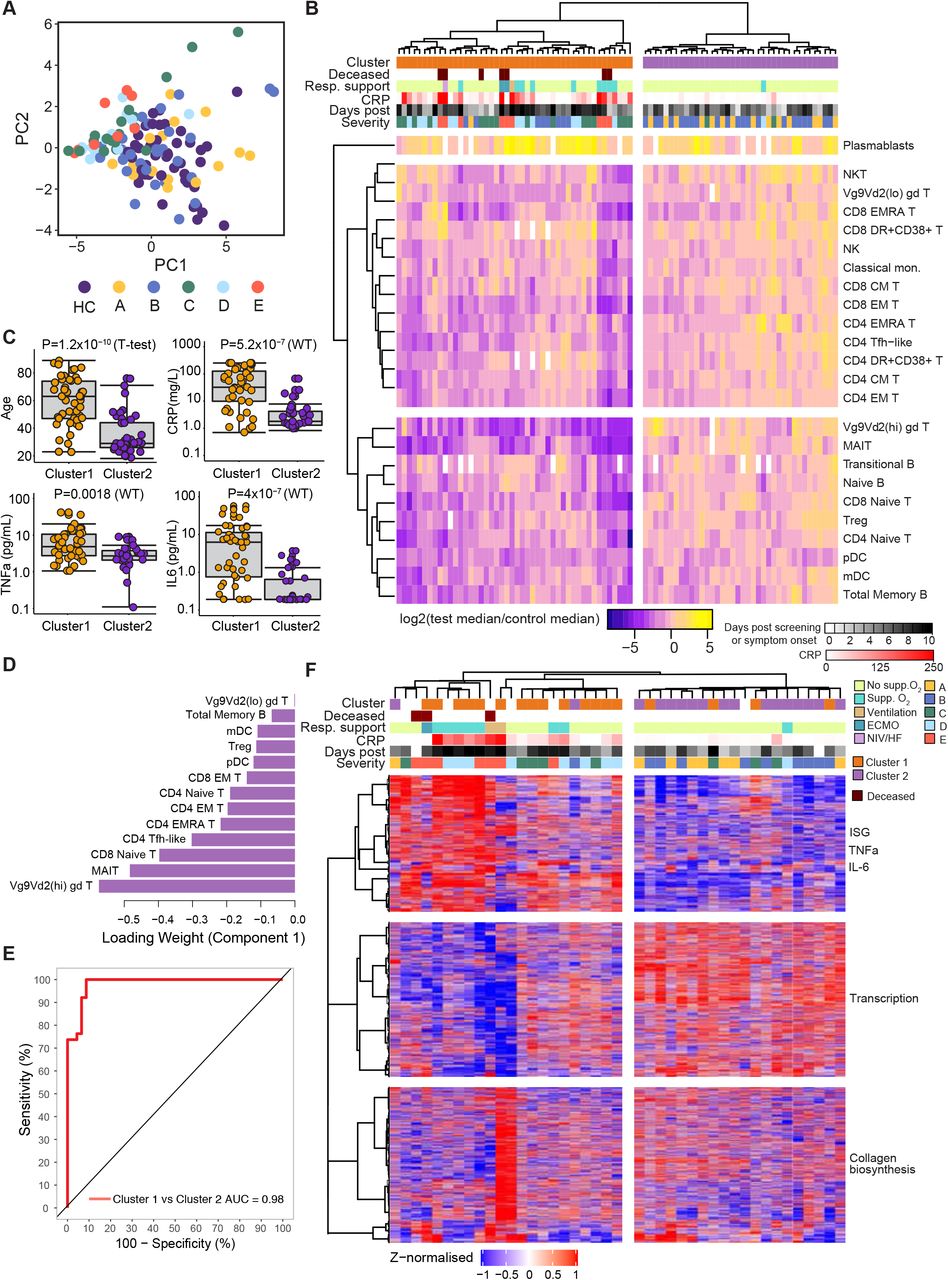
A) Principal component analysis of peripheral blood absolute cell counts for 24 key cell subsets from HC and COVID-19 cases, for samples taken <10 days from screening (group A) or symptom onset (group B-E). Points are coloured according to severity category. B) Unsupervised clustering of absolute cell counts (normalised to the median of healthy controls) for COVID-19 samples and cell types included in A), showing grouping of cases into two clusters (cluster 1, orange; cluster 2, purple) by Euclidean distance and Ward D hierarchical clustering. C) Boxplots comparing age and inflammatory characteristics of individuals in clusters 1 and 2 at the time of sampling. D) Thirteen cell types selected by sPLS-DA as most informative in predictive models discriminating patients in clusters 1 and 2. Bars indicate loading coefficient weights of selected features (ranked from most to least informative in cluster prediction, from bottom to top). E) AUROC curve showing sensitivity of cluster group prediction at varying specificity thresholds, based on absolute counts of 13 selected cell types. F) K-means clustering of 18357 whole blood transcripts from COVID-19 samples taken <10 days fromscreening or symptom onset. Gene clusters are annotated for enriched signatures, samples are annotated according to corresponding cluster membership in B) where possible.
Thus, it appears that abnormalities in perhaps 13 cell subsets, when detected at presentation with the symptoms of COVID-19, define a group with, or who progress to, severe COVID-19 disease. Twenty of 36 patients within cluster 1 required respiratory support, and this was already maximal at initial blood sampling in 14 of them. In 6 patients, membership of this group was thus predictive of treatment escalation, in another 7 it was predictive of death – so clustering by immunophenotype correlated with severity in essentially all patients, and with subsequent disease progression in 36%. Such an observation, once independently validated and correlated with other indicators of disease severity (such as imaging, oxygen saturation measurements, and CRP and other inflammatory markers), could form part of clinically useful prognostic assessment tools.
Characteristics of a successful anti-SARS-CoV-2 immune response
HCW in categories A and B did not progress to severe COVID-19 disease, and they also cluster apart from those with more severe disease when using either early immune cell counts or RNA expression profiles (Figure 4). We therefore compared the early features of these successful immune responses to those in patients with more severe COVID-19, to shed light on what differentiated effective antiviral responses from those associated with progression to severe disease - binned now in 7-day intervals to provide finer definition of the earlier immune response. For comparison, we show CRP and selected complement components (Figure 5A).

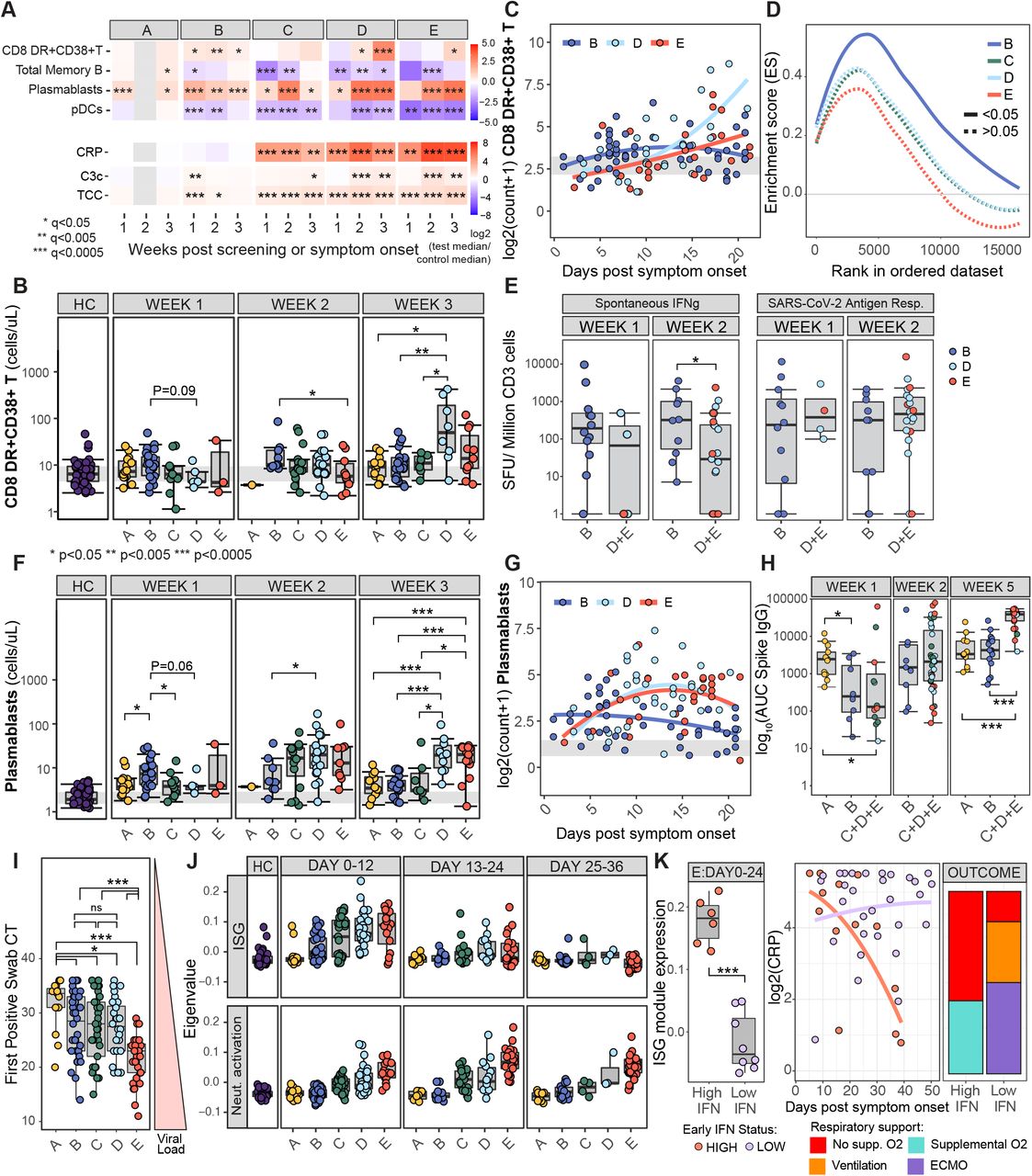
A) Heatmap showing the log2 fold change in median absolute cell counts, CRP or complement measures between COVID-19 cases and HCs, within severity categories and across 7-day time bins from screening (group A) or symptom onset (group B-E). Wilcoxon test FDR adjusted p-value: *<0.05, **<0.005, ***<0.0005. B) Boxplots showing absolute non-naive HLA-DR+CD38+ CD8 T cell counts (cells/uL) across severity categories from weeks 1-3 post screening or symptom onset. Wilcoxon tests p-values annotated as above. Grey bar represents the interquartile range of the same population in HCs. C) Mixed-effect model with quadratic time trend showing corresponding cellular trajectories in sample group B,D and E. D) Enrichment score for CD8 T-cell activation signature as determined by GSEA in group B-E for samples taken <24 days from symptom onset. E) Number of CD3 T cells secreting IFN γ spontaneously or following SARS-CoV-2 antigen stimulation, in patient samples from groups B, and D and E combined, one or two weeks post symptom onset. Kruskal-Wallis test p-values annotated as in B). Equivalent boxplot and longitudinal modelling of plasmablast counts are shown in F) and G). H) Area under the curve for SARS-CoV-2 spike-specific IgG titres at 1, 2 and 5 weeks post screening (group A) or symptom onset (group B-E). Groups C,D and E are combined for increase statistical power, p-values as in E. I) Boxplots showing SARS-Cov-2 viral load, taken as first positive swab PCR cycle threshold (CT), in severity groups. Wilcoxon test P-values annotated as in B. J) Boxplotscapturing expression of ISG and neutrophil activation-related transcriptomic eigenvalues across disease severity and time. K) Stratification of group E samples taken <24 days post symptom onset into high and low expression of interferon stimulated genes (ISG), with persisting and resolving CRP status and final respiratory status reported within 12 weeks shown by bar charts. SFU, spot forming units
A number of key features emerge. First, there is no evidence of systemic inflammation in groups A and B. CRP is normal (Figure 5A), cytokines are not raised (Figure 1E), and there is no RNA evidence of systemic inflammatory gene-related signatures (Figures 3B and 3C). The exception is the significant but transient increase in C3c and TCC. Most cell types which are profoundly reduced in groups C to E are normal in A and B (Figure 2), but some show mild reductions in group B in particular, of which we show pDCs and memory B cells as examples (Figure 5A). pDCs fall to a lesser extent than in more severe disease, and for a shorter time period; a fall that is perhaps consistent with tissue localisation to allow local interferon production as part of a successful antiviral response. There is an early increase in cytotoxic CD8 T cells seen in group B compared to the rise seen in groups C to E, with the increase seen by day 7 and peaking up to 2 weeks after symptom onset. This contrasts with the later and more sustained rise seen in the more severe COVID-19 patients (Figures 5A, 5B and 5C). There is enrichment of a CD8 cytotoxic RNA signature in group B by GSEA, which is significantly raised compared to group C to E between 0 and 24 days after symptom onset (Figure 5D). Consistent with these findings, spontaneous generation of IFNγ by T cells, as detected by ELISpot analyses, is more pronounced in group B at samples taken two weeks post symptom onset (Figure 5E). A robust anti-SARS-CoV-2-specific T cell response is also seen in group B, comparable to more severe groups (Figure 5E and S6). There is also an early increase in plasmablasts seen in both groups A and B, again occurring up to a week before a larger rise is seen in more severe disease (Figures 5F and 5G). Anti-Spike IgG is also prominent in groups A and B at weeks 1 and 2 (though samples from asymptomatic patients in group A are timed since their swab, and may be further out from infection than those able to be timed from symptom onset) (Figure 5H). Taken together, these data suggest that an early adaptive immune response is prominent in individuals who are asymptomatic or have mild disease, characterised by a rapid production of cytotoxic CD8 T cells, plasmablasts and likely pDC tissue localisation.
Virus at first swab, as assessed by PCR CT value, is comparable in groups B, C and D, and is low in group A (again perhaps in part because these samples may be taken later after infection than symptomatic groups, see above). Initial viral titre was therefore not associated with an increased risk of hospital admission (being similar in groups B, C and D), but was significantly higher in group E than in other groups (Figure 5I). These viral titres are reflected in interferon-related transcription signatures, which are prominent in groups B-E (Figure 5J). If the strength of this early antiviral response helps govern outcome, it might be that a higher initial interferon response is associated with a better prognosis. That those in group E with low interferon signatures in early disease are more likely to have persistently high CRP appears consistent with this notion (Figure 5K), though this needs confirmation in a larger dataset.
In those with more severe disease (groups C-E), evidence of systemic inflammation is present from the first blood test, evident in samples taken from 0 and 7 days from the onset of symptoms (Figure 5) but evident early in that window. If we focus on the 16 patients in groups C-E sampled between 2 days before and 4 days after symptom onset, 15 had a CRP > 10 and/or neutrophil activation eigengene > 0. All of the 5 patients sampled between 2 days before and 2 days after symptom onset met these criteria. Thus, inflammation does not develop later from the progression of a non-inflammatory immune response or as a result of failure to clear virus. The immunopathology associated with COVID-19 appears to develop at or very early after infection, making strategies to prevent its onset difficult.
Distinct patterns of immune recovery in COVID-19
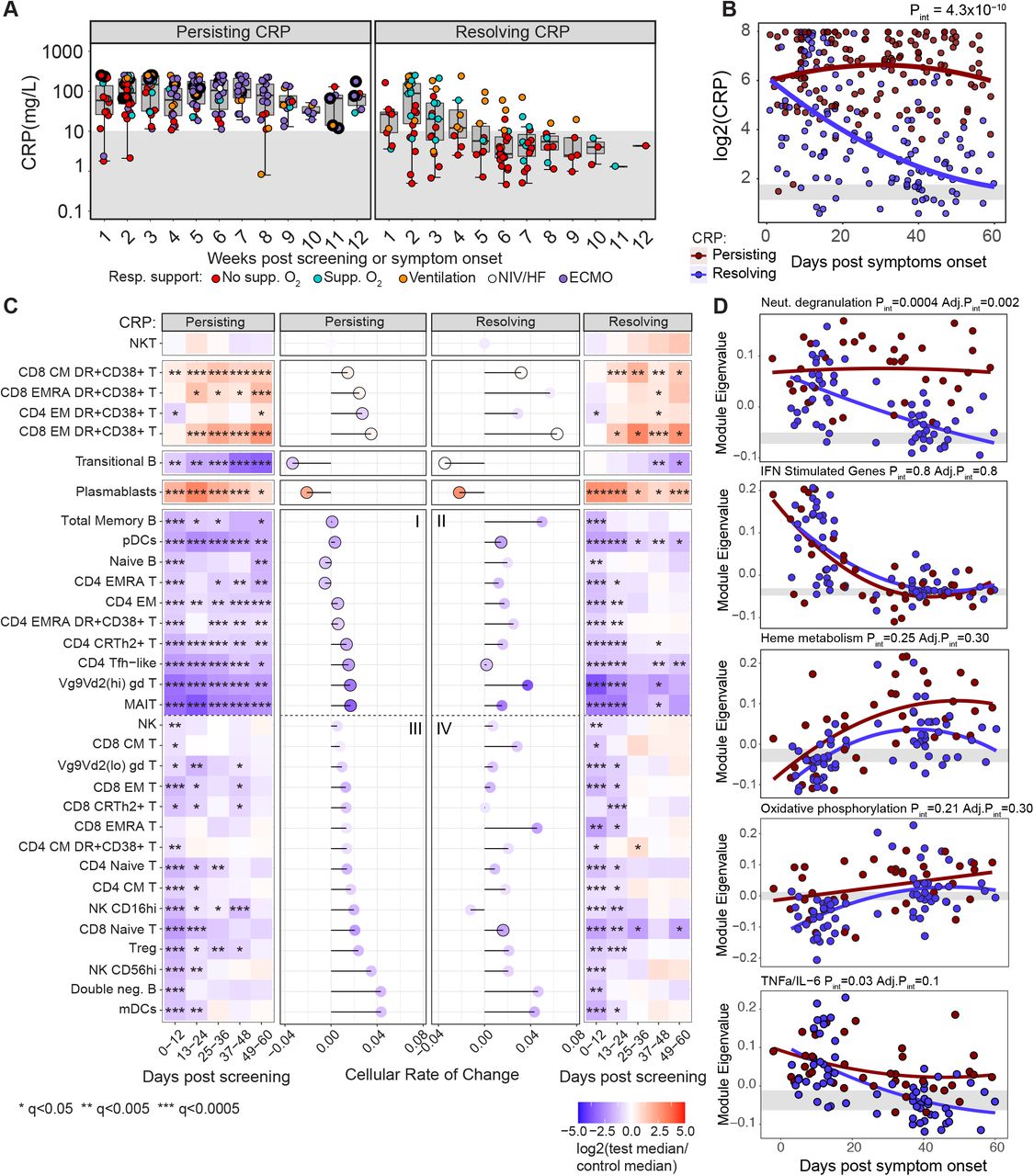
In contrast to groups A and B, cellular changes in groups C - E were profound and usually most prominent at the first bleed (Figure 2). Determination of the rate and direction of change among immune cell subsets over time was likely to be most informative in these groups, and we therefore explored immune cell kinetics in groups C, D and E, assigning patients to two categories based on whether their CRP levels remained elevated above 10mg/L (“Persisting CRP”) or fell below 10mg/L (“Resolving CRP”) by their final bleed within 3 months post symptom onset (Figure 6A). The latter group included both individuals with high CRP levels early, and those for which CRP remained low (10mg/L) at all measured time points over the course of study. Changes in CRP over time differed significantly between these two groups when assessed using a mixed-effects model, with time modelled as a continuous variable (Figure 6B).

A) CRP (mg/L) over 7-day time bins in patients grouped by persisting and resolving CRP. B) Mixed-effect model with quadratic time trend showing log2(CRP) trajectories in both patient groups, the likelihood-ratio test p-value for the time x group interaction term is reported. Grey band indicates the interquartile range of the corresponding measure in HCs. C) Heatmap showing the log2 fold change in median absolute cell count between COVID-19 cases and HCs, within CRP groups and across 12-day time bins. Wilcoxon test FDR adjusted p-value: *<0.05, **<0.005, ***<0.0005. Rate of cellular change is shown for each cell by lollipop plot; a faster rate of recovery, or deviation from normal, is indicated by increasing stem length. Points are coloured by log2 fold change in median absolute cell counts from HC at 0-12 days, black outline indicates failure to recover to HC levels within 60 days (as defined in methods). D) Mixed-effect models showing longitudinal trajectories of gene module eigenvalues capturing neutrophil degranulation, interferon stimulated genes (ISG), heme metabolism and oxidative phosphorylation in CRP groups, p-values reported as previously.
In order to compare the nature of cellular changes over time, both across cell subsets and between persisting and resolving CRP patient groups, a “rate of change” for each cell population was calculated over 60 days post symptom onset. In brief, this rate captured both the initial deviation in cell counts from normal within a window of 0-12 days, and the time taken for cells counts to stabilise within a normal range if cellular recovery did occur (see Methods). Five predominant trajectories were observed; populations that did not deviate from heathy levels over the duration of study (e.g. NKT cells), those which increased progressively from normal over time (e.g. effector CD8 T cells), those which fell progressively from normal over time (e.g. transitional B cells), those which trended toward recovery after an initial rise in numbers (plasmablasts) and those which tended toward recovery after an initial drop in numbers (e.g. naïve CD4 T cells) (Figure 6C).
The absolute number of most cell populations fell precipitously early, and then showed variable degrees of recovery. For descriptive purposes these are arranged into a group of cell subtypes that failed to recover, or recovered, in the persisting CRP group (Figure 6C quadrants I and III respectively), and their equivalents in the resolving CRP group (Figure 6C quadrants II and IV respectively). Notably, a few populations remained consistently abnormal in both persisting and resolving CRP groups out to 60 days post symptom onset, including pDCs, Tfh, MAIT cells and Vg9Vd2 (hi) γδ T cells. All other populations showing an early drop in counts (with the exception of naïve CD8 T cells) recovered to normal levels in the patients with resolving CRP (II and IV), and at rates more rapid than seen in those with persisting high CRP values. In the persisting CRP group, a number of cell types remained markedly abnormal (including memory B cells and various CD4 T cell subsets: quadrant I), whereas a second group of cell types recovered despite persisting inflammation (including NK cells and a number of CD8 T-cell subsets: quadrant III).
We then explored the relationship between cell recovery and the nature and kinetics of the inflammatory response. It is not surprising that where the CRP remains persistently elevated, immune defects might persist, on the assumption that these defects are secondary to the inflammatory state. Consistent with this, the cohort with persistently raised CRP also has raised TNF-α and IL-6 at the protein level over time (Figure 1D). Likewise, transcriptional signatures of TNF-α, IL-6 and neutrophil activation were increased in severe disease (Figure 3), particularly in the persistent CRP group (Figure 6D). This ongoing inflammation may contribute to the sustained reduction in cell numbers at late times seen in quadrant I, together with persistently raised HLA-DR+/CD38+ effector T cells and plasmablasts (Figure 6C). Consistent with this, it is also perhaps not unexpected that most cell types reduced in acute disease recover over a few weeks as the CRP falls, as is seen for most cells in quadrants II and IV (Figure 6C).
More intriguing are the cells that recover rapidly in the face of ongoing inflammation (quadrant III). While the reasons for this are likely to differ between cell types, and to be multifactorial, there is the possibility that some of these cell reductions are driven by the viral infection per se, virus-induced interferon, or a combination of the two. It is notable that, after an initial rise, IFN-γ returns to normal in patients irrespective of disease severity (Figures 1D and 1E). Interferon-stimulated gene (ISG) signatures fall to normal levels over about 3 weeks independent of disease severity group (Figure 3C) and CRP (Figure 6D), but correlating with declining virus titre (Figures 3D and S4E). Thus, cell types known to leave the circulation due to interferon stimulation (Kamphuis et al., 2006), such as T and NK cells (Hirsch and Johnson, 1986; Zafranskaya et al., 2007), may recover as interferon-dependent inflammation falls, presumably as a result of control of viral infection and independently of ongoing CRP-associated inflammation.
Finally, a small number of cell types remain statistically abnormal after 60 days, even in the resolving CRP group. These include effector CD4 and CD8 T cells (HLA-DR+/CD38+), and plasmablasts, all of which remain elevated, and pDCs, Tfh, Vg9Vd2 expressing γd T cells and MAIT cells, which remain reduced (Figure 6C) and are among those cells most predictive of poor prognosis (Figure 4D). These abnormalities persist despite resolution of CRP-reflected inflammation, with evidence for neutrophil degranulation, TNF-α /IL-6 and glycolysis all falling alongside CRP (e.g. Figures 1D and 6D). They also persist despite early resolution of interferon-stimulated gene signatures in all severity groups. Possible mechanisms behind these sustained abnormalities are discussed below.
The late appearance of OXPHOS and ROS pathways correlates with differential immune recovery
At late time points, whole blood transcriptome analysis shows an increase in inflammation-related signatures distinct from those that are prominent early in the disease course, particularly in severity groups C-E. These signatures are characteristic of oxidative phosphorylation (OXPHOS), reactive oxygen species generation (ROS) and heme metabolism. These are demonstrated in an un-biased fashion using WGCNA, where modules characterised by OXPHOS and heme metabolism signatures are prominent in samples analysed at day 25 to 48 post symptoms, with OXPHOS most prominent in group E, and heme metabolism in C, D and E (Figures 3B and 3C). Enrichment of Hallmark signatures in RNA-seq datasets confirmed the association of OXPHOS and heme metabolism in groups C, D and E, and also found association of a ROS signature (Figures 3E and 7A). Consistent with this, the expression of the genes driving the enrichment of each signature was upregulated in the three most severe clinical groups (Figure 7B and Table S3). The late rise in these three correlated signatures occurs irrespective of persisting or resolving CRP-associated inflammation (Figure 6D).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A) Enrichment score for HALLMARK genesets capturing heme metabolism, oxidative phosphorylation and ROS related genes (as determined by GSEA) in group A-E samples taken 25-48 days post screening or symptom onset. B) Heatmap showing enrichment for the intersection of GSEA leading edge genes from groups C,D and E, across all severity groups in samples taken 25-48 days post screening or symptom onset. C) Heatmap showing correlation between transcriptional eigengenes and absolute cell counts, at 25-48days post symptom onset. Boxes are coloured by strength of correlation, Pearson correlation p-values: *<0.05, **<0.01, ***<0.001. D) Schematic illustrating the concentration of profound changes seen at the first blood sample at around the time of symptom onset, truncating a process suggested by some to be sequential to associations that can all be made from a single blood sample at around the ime of symptom onset. E) A schematic showing the trajectory of viral titre (grey) and inflammatory transcriptional signatures (yellow, including TNF/IL-6, OXPHOS, ROS and interferon-stimulated genes, ISG) in COVID-19 patients with persistent or resolving CRP (red).
We then sought correlation between cellular and transcript signature changes in COVID-19. In the first 24 days after symptom onset, there is a strong association between TNF-α /IL-6, neutrophil degranulation and interferon signatures with most of the lymphoid cell types whose numbers fall in severe disease (Figure S7). Later, between 24 and 48 days after symptom onset, these associations change (Figure 7C). While TNF-α /IL-6 and neutrophil degranulation signatures are still associated with many cell subsets that continue to be reduced, the interferon signature is no longer a significant player. Strikingly, the persistent increase seen in effector lymphocytes, both CD4 and CD8 activated T cells (HLA-DR+, CD38+) and plasmablasts, is now particularly associated with the OXPHOS signature which, having become more prominent later in disease (Figure 3B), has a much more restricted and specific association with immune dysfunction than other inflammatory signatures.
It is thus clear that, for some cell types, the association with the inflammatory milieu changes over time, but for others it is more consistent. It is interestingly the inflammatory signatures which appear late in disease, in particular OXPHOS, are specifically associated with persistent derangement of cell types of potential pathological importance, such as increased HLA-DR+CD38+ T cells and plasmablasts, and reduced pDCs.
Discussion
This study has been able to compare very early immune responses in SARS-CoV-2 infected individuals whose disease is asymptomatic or mildly symptomatic, and does not progress (groups A and B respectively), to those in which more severe COVID-19 manifestations become apparent (groups C-E). In groups A and B there is evidence of an early robust adaptive immune response. This is characterised by an increase in circulating plasmablasts, and in CD8+HLA-DR+CD38+ activated T cells, which expand earlier and in higher numbers than in more severe COVID-19 groups, most notably in the first week after symptom onset. Both of these cell populations then contract in A and B, as they continue to rise in groups C-E.
Consistent with this there is prominent early anti-spike antibody in patients in groups A and B. Total activated T cells are increased, as demonstrated by anti-interferon gamma ELISpots performed immediately ex vivo, and robust early SARS-CoV-2 specific T cell responses are also apparent. These early cellular changes are accompanied by a prominent early interferon signature. Others have shown that anti-SARS-CoV-2 responses can be seen early in disease (Rydyznski Moderbacher et al., 2020), though their prominence in asymptomatic and very mildly symptomatic individuals has not been established (e.g. only 2 patients in (Rydyznski Moderbacher et al., 2020) would have fallen within groups A and B of our classification).
At the same time as these signs of an early adaptive immune response are seen in groups A and B, there is no evidence of systemic inflammation, apart from some early, transient complement activation. CRP, circulating TNF-α and IL-6, and transcriptional signatures of a number of inflammatory pathways (including those associated with neutrophil activation and TNF-α /IL6 signalling) are not raised in groups A and B, at time points when they are already prominent in groups C-E. The severe and widespread leucocyte subset depletion seen at the initial bleed in the more severe COVID-19 patient groups C-E, and observed by many others (Carissimo et al., 2020; Laing et al., 2020; Mathew et al., 2020; Su et al., 2020), is not apparent in those with asymptomatic or mildly symptomatic disease, suggesting this is a unique feature of a pathological immune response. Absolute cell counts from 13 immune cell subsets measured within 10 days of symptom onset can identify patients with severe and progressive disease. Coupled with evidence of early systemic inflammation seen in severity groups C-E, our findings suggest that the immune pathology associated with severe COVID-19 is either established immediately post-infection or, if there is a transition point from an effective to a pathological response, this is likely to occur before or around the time of symptom onset (Figure 7D). This finding may have major implications as to how disease needs to be managed, since intervention to prevent immune pathology would need to be targeted very early in the disease course, and perhaps pre-emptively in high risk groups screened and diagnosed before symptoms develop.
The reason for the failure to mount a robust early B and T cell response in the context of severe COVID-19 is likely to be multifactorial. There is no evidence for a relationship with viral load and progression to inflammatory disease, as initial viral titres were comparable between group B and groups C and D. Once inflammatory disease is established, however, viral titre may be associated with subsequent outcome (as early increased viral titre is seen in group E), consistent with reports that high initial viral titre might be associated with mortality (Pujadas et al., 2020). Genetic association studies in severe COVID-19 point to genes that are implicated in driving antiviral responses, such as those involved in type 1 interferon-mediated immunity (Pairo-Castineira et al., 2020; Zhang et al., 2020), and increasing age and comorbidity such as diabetes and chronic inflammatory disease are all known to suppress early CD8 T cell and B cell responses (Shen-Orr et al., 2016; Weiskopf et al., 2009). An in-depth understanding of these risk factors may instruct screening strategies to assess risk of progression before inflammatory responses become self-sustained.
While clear distinctions in immune responsiveness were apparent between groups A and B versus groups C, D and E, differences between the more severe groups themselves were less obvious. Those with symptomatic disease warranting admission to hospital clustered together using the size of just 13 key cell populations: these clusters correlated strongly with clinical severity, and provided prediction of subsequent progression, as well as COVID-19 associated death. Similar observations have been made by others (Chen and Wherry, 2020), though whether the predictive ability of clustering by immune parameters adds to that provided by clinical and other predictors remains to be determined. While determining prognosis after presentation to hospital could be of clinical use, it would be of more benefit to predict progression to severe disease in cases with milder COVID-19 – but it is not clear whether this will be possible in practice, given our observation that inflammatory immunopathology is already present at first presentation. A study to address this issue would need to be conducted in particularly high-risk patient groups to ensure an adequate event rate, and require diagnosis through asymptomatic screening to detect changes before symptoms develop.
The recovery of the profound immune dysregulation seen in those with severe COVID-19 is potentially of major clinical relevance, as such recovery may be required to prevent secondary infection or SARS-CoV-2 reinfection, and persistent immunopathology could be associated with clinical manifestations of “long COVID”. We show that profound alterations in many immune cell types often persist for weeks to months after SARS-CoV-2 infection, and different cell populations exhibit strikingly different patterns of resolution. Some recover as systemic inflammation (as measured by CRP) resolves, others remain persistently abnormal despite a drop in CRP toward normal levels, and a third group resolve even in the face of persistent systemic inflammation.
Understanding the different inflammatory drivers or associations of this differential recovery could provide insight into the immune pathology of COVID-19, and potentially of other infections. Here, to begin to explore this, we correlated immune changes with measurements of systemic inflammation throughout the disease course. Patients with severe COVID are characterised by high CRP, and this correlates with evidence of TNF-α and IL-6 driven processes both at the protein and transcriptome level, as well as with both neutrophil activation and glycolytic metabolism. The fact that many cellular abnormalities persist while these biologic processes are apparent – while others appear to resolve alongside them – suggests that the nature of systemic inflammation is important in driving different aspects of immune pathology.
Finally, some cell populations remain markedly abnormal, or show a limited recovery, even once CRP-associated inflammation has resolved, and indeed after patients have been discharged from hospital. These persistent changes may reflect a slow intrinsic regenerative capacity of the cell type concerned, but in other situations, such as the continued elevation of effector T and B cells, it is tempting to speculate that there is ongoing abnormal signalling driving such changes. For that reason, we explored late changes that are seen in the inflammatory response in COVID-19.
Interestingly, three transcriptional signatures arise late in those with severe COVID-19 and are not present in early severe, nor mild, disease. These include activation of OXPHOS-, ROS- and heme-related metabolic pathways (Figure 7E). Activation of immune cells results in metabolic reprogramming that supports cell growth, proliferation and differentiation. Disruption of metabolic pathways can result in bioenergetic, anabolic, epigenetic or redox cellular crises – culminating in immune dysfunction (Bantug et al., 2018). It is unlikely that the metabolic signatures observed here simply reflect heightened bioenergetic requirements of activated immune cells, as one would expect that similar requirements are present also at early stages in the disease. OXPHOS can drive inflammation (Mills et al., 2017), and it is intriguing to note COVID-19 patients treated with metformin, which inhibits Complex I of the respiratory chain, had lower amounts of circulating inflammatory cytokines (Cheng et al., 2020). The ROS transcriptional signature may relate to more abundant production of ROS-species inevitably accompanying increased OXPHOS. Alternatively, it may reflect specific mitochondrial pathology, and thus per se contribute to immune cell dysfunction (Nathan and Cunningham-Bussel, 2013). Mitochondria are also critically involved in heme biosynthesis. Heme serves as a prosthetic group for haemoglobin as well as many other proteins – including several that constitute the respiratory chain of mitochondria. While free heme can act as damage-associated molecular pattern and promote ROS formation, the role of heme biosynthesis vs. catabolism in balancing cellular sensitivity to oxidants is complex and context dependent (Prestes et al., 2020). Here, given correlated regulation of heme and OXPHOS pathways in the clinical categories C, D and E, activity of these modules may be interrelated and possibly jointly reflective of dysfunctional mitochondria. How heme and OXPHOS transcriptional programmes are linked on a molecular level cannot be inferred from our data. Erythroid cell activation has recently been detected in severe COVID-19 (Bernardes et al., 2020) and could also contributes to a heme transcriptional signature. However, the increase in heme metabolism in our cohort correlates strongly with a falling haemoglobin, and reticulocytes (Figure S7) in patients in groups C, D and E are low – suggesting suppression rather than activation of erythropoiesis in these individuals.
Understanding the mechanism linking metabolic dysregulation to persistent immune pathology in COVID-19, and also to “long COVID”, will require further study over longer disease courses.
Author Contributions
Conceptualization: C.H., J.R.B., P.A.L., and K.G.C.S.; Methodology: L.B., L.T., M.R.W., I.G.G., and R.D.; Software: K.H.; Validation: L.T.; Formal Analysis: L.B., F.M., L.T., A.H., P.K., and H.R.; Data Curation: F.M., A.H., P.K., L.T.; Investigation: L.B., F.M., L.T., P.K., A.D.S., O.H., M.R.W., I.G.G., M.H., R.D., J.K.N.,and E.J.M.T.; Visualization: A.H., and P.K; Project Administration: L.B., F.M., L.T., B.J.D, C.S. and A.E.; Funding Acquisition: P.J.L., N.K., J.R.B., C.H., G.D., M.P.W., P.A.L, and K.G.C.S.; Supervision: J.R.B,P.A.L and K.G.C.S.; Resources: A.M.P., M.T., M.P.W., N.J.M.; Writing – Original Draft: P.A.L, P.J.L., C.H. and K.G.C.S; Writing – Review & Editing: L.B., F.M., A.H., L.T., P.K., H.R., B.J.D., A.D.S., O.H., R.D., K.H., S.B., R.K.G., J.K.N., C.S., A.E., A.M.P., E.J.M.T., N.K., P.J.L., N.J.M., S.R., J.E.D.T., M.P.W., M.T., C.H., J.R.B, G.D., M.R.W., M.H., I.G.G., P.A.L., and K.G.C.S.
Declaration of Interests
The authors declare they have no competing interests.
Methods
Participant recruitment
Study participants were recruited between 31/3/2020 and 20/7/2020 from patients attending Addenbrooke’s Hospital with a suspected or nucleic acid amplification test (NAAT) confirmed diagnosis of COVID-19 (including point of care testing (Collier et al., 2020; Mlcochova et al., 2020)), patients admitted to Royal Papworth Hospital NHS Foundation Trust or Cambridge and Peterborough Foundation Trust with a confirmed diagnosis of COVID-19, together with Health Care Workers identified through staff screening as PCR positive for SARS-CoV-2 (Rivett et al., 2020). Controls were recruited among hospital staff attending Addenbrooke’s serology screening programme, and selected to cover the whole age spectrum of COVID-19 positive study participants, across both genders. Only controls with negative serology results (45 out of 47) were subsequently included in the study. Recruitment of inpatients at Addenbrooke’s Hospital and Health Care Workers was undertaken by the NIHR Cambridge Clinical Research Facility outreach team and the NIHR BioResource research nurse team. Ethical approval was obtained from the East of England – Cambridge Central Research Ethics Committee (“NIHR BioResource” REC ref 17/EE/0025, and “Genetic variation AND Altered Leucocyte Function in health and disease - GANDALF” REC ref 08/H0308/176). All participants provided informed consent.
Inpatients were sampled at study entry, and then at regular intervals as long as they remained admitted to hospital (approximately weekly up to 4 weeks, and then every 2 weeks up to 12 weeks). Discharged patients were invited to provide a follow-up sample 4-8 weeks after study enrolment. Health care workers were sampled at study entry, and subsequently after approximately 2 and 4 weeks. At each time-point, blood samples were drawn in EDTA, sodium citrate, serum and PAXgene Blood RNA tubes (BD Biosciences).
Clinical data collection
Clinical data were retrospectively collected by review of medical charts and entered into spreadsheets or Castor EDC, a cloud-based clinical data management system. Available laboratory test results and administered in-patient medications were extracted from Epic electronic health records (Addenbrooke’s Hospital) and from MetaVision ICU (RPH ITU). Data were merged from the various data sources using R version 3.6 and the R packages readr (1.3.1), openxlsx (4.1.4), dplyr (0.8.3), tidyr (1.0.2) and lubridate (1.7.4).
Health care workers were classified in 2 groups (A and B) according to whether they were asymptomatic (group A) or had possible COVID-19 symptoms (group B) at the time of PCR testing. For this purpose, new-onset fever (>37.8 C), cough, loss of sense of smell, hoarseness, nasal discharge or congestion, shortness of breath, wheeze, headache, muscle aches, nausea, vomiting and diarrhoea were considered as possible COVID-19 symptoms.
Participants in group A were further sub-grouped according to whether they were completely asymptomatic (n= 8), or had had any of the above COVID-19 symptoms before PCR testing (n=10, median time from symptoms to COVID-19 PCR test 26 days, range 9-42 days).
Group B participants included both staff who were self-isolating because of COVID-19 symptoms (n=30), and staff members who were reporting fit for duty, but had some symptoms that did not reach the threshold for self-isolation at that time (n=10).
Hospital patients were assigned to 3 severity groups, mainly reflecting the maximum level of respiratory support for COVID-19 received during their hospital stay:
- group C: did not receive any supplemental oxygen. Five patients were discharged soon after initial diagnosis and assessment but followed up as part of the study.
- group D: received supplemental oxygen using low flow nasal prongs, simple face mask, Venturi mask or non re-breather face mask
- group E: received any of non-invasive ventilation (NIV), mechanical ventilation or ECMO. Patients who received supplemental oxygen (but no ventilation) and deceased in hospital were also assigned to group E.
In patients who were already established on home NIV for chronic respiratory failure, NIV delivered as per the home prescription (e.g. nocturnal) was not considered for the purpose of classification. Moreover, oxygen requirements that were clearly not related to COVID-19 were also not considered for classification purposes. In particular, 2 cases who received low flow supplemental oxygen for non-COVID-19 indications (ascitic splinting in decompensated cirrhosis in one case, and recovery from anesthesia after orthopedic surgery in the other) were assigned to group C. Cases in group C were further sub-classified according to chest radiology results (X-ray and, if available, CT scan), as:
- abnormal radiology: chest X-ray/ CT scan showed changes compatible with COVID-19
- normal radiology: chest X-ray/ CT scan did not show any abnormality compatible with COVID-19 (reported as normal or showing lung changes diagnostic of conditions other than COVID-19).
Immunological parameters were analyzed according to time since onset of symptoms, or otherwise time since positive SARS-CoV-2 NAAT (in group A and in 4 asymptomatic patients in group C). Seven cases admitted to hospital for COVID-19 had no date of onset of symptoms documented in the medical records. In these cases, the date of onset of symptoms was estimated as follows: hospital admission date - median time from symptoms to hospital admission in patients admitted for COVID-19.
Following clinician review, 6 cases were considered not classifiable, due to complex concomitant pathologies that coexisted with COVID-19 and dominated the clinical picture, confounding the interpretation of clinical outcome. These cases were not included in any analyses; more details are reported in Table S2. Figure S1 summarizes the timing of research samples and clinical trajectories for volunteers in severity groups C, D and E included in the analysis.
Peripheral blood mononuclear cell preparation and flow immunophenotyping
Each participant provided 27 mL of peripheral venous blood collected into 9 mL sodium citrate tube. Peripheral blood mononuclear cells (PBMCs) were isolated using Leucosep tubes (Greiner Bio-One) with Histopaque 1077 (Sigma) by centrifugation at 800x g for 15 minutes at room temperature. PBMCs at the interface were collected, rinsed twice with autoMACS running buffer (Miltenyi Biotech) and cryopreserved in FBS with 10% DMSO. All samples were processed within 4 hours of collection.
Five distinct antibody cocktails (Table S4) were used to label approximately 106 PBMCs using standard methods. T regulatory cells were fixed and permeabilised following surface staining prior to the addition of intracellular antibodies. Samples were stored at 4°C and acquired within 4 hours using a 5-laser BD Symphony X-50 flow cytometer. Single colour compensation tubes (BD CompBeads) or cells were prepared for each of the fluorophores used and acquired at the start of each flow cytometer run.
For direct enumeration of T, B and NK cells, an aliquot of whole blood (50μl) was added to BD TruCount™ tubes with 20μl BD Multitest™ 6-colour TBNK reagent (BD Biosciences) and processed as per the manufacturer’s instructions.
Samples were gated in FlowJo v10.2 according to the schema set out in Figure S8. The number of cells falling within each gate was recorded. For analysis, these were expressed as an absolute concentration of cells per μl, calculated using the proportions of daughter populations present within the parent population determined using the BD TruCount™ system.
CyTOF
The protocol used to isolate PBMCs led to an impaired recovery of the different monocytes population, specifically intermediate and non-classical monocytes. To extend our analysis to these and other granolucyte populations we performed mass cytometric analysis on a subgroup of patients and healthy controls (249 samples). Briefly, whole blood samples (270μl) were stained using the Fluidigm Maxpar® Direct™ Immune Profiling Assay™ according to the manufacturer’s instructions. Samples were cryopreserved at −80°C following staining and thawed for analysis within 4 weeks. Samples were acquired using a Fluidigm Helios mass cytometer and normalised using the CyTOF Software v6.7.1016. FCS files generated were analysed using the Maxpar® Pathsetter™ software v2.0.45 (Verity Software House, Topsham, ME). Standard settings were used to generate immune cell frequencies for 37 immune cell populations. Absolute cell numbers were calculated using the proportions of these immune cell populations within the parent populations determined by BD TruCount™.
Reticulocyte counts
Reticulocyte numbers were measured using a Sysmex XN-1000 haematology analyser as previously described (Akbari et al., 2020).
Complement
Complement activation was assessed by measuring C3 activation products (C3a and C3c) together with the terminal complement complex (TCC) as an end product of the complement cascade.
Concentrations of these complement components were measured in EDTA plasma from patients using commercially available enzyme-linked immunosorbent assays (ELISA) kits (HK354 (C3a), HK368 (C3c), HK328 (TCC), Hycult Biotech, Uden, the Netherlands) according to the manufacturer’s protocols.
CRP
High sensitivity CRP was measured using the standard assay by the Core Biochemical Assay Laboratory (CBAL) at Cambridge University Hospitals NHS Foundation Trust.
Cytokines
IL-6, IL-10, IL-1β, TNFα and IFNγ were measured using standard clinical assays performed by the Clinical Immunology Laboratory at the Department of Biochemistry and Immunology, Addenbrooke’s Hospital Cambridge.
IFNγ FLUOROSPOT assays
Frozen PBMCs were rapidly thawed, and the freezing medium was diluted into 10ml of TexMACS media (Miltenyi Biotech), centrifuged and resuspended in 10ml of fresh media with 10U/ml DNase (Benzonase, Merck-Millipore via Sigma-Aldrich), PBMCs were incubated at 37°C for 1h, followed by centrifugation and resuspension in fresh media supplemented with 5% Human AB serum (Sigma Aldrich) before being counted. PBMCs were stained with 2ul of each antibody: anti-CD3-fluorescein isothiocyanate (FITC), clone UCHT1; anti-CD4-phycoerythrin (PE), clone RPA-T4; anti-CD8a-peridinin-chlorophyll protein - cyanine 5.5 (PerCP Cy5.5), clone RPA-8a (all BioLegend, London, UK), LIVE/DEAD Fixable Far Red Dead Cell Stain Kit (Thermo Fisher Scientific). PBMC phenotyping was performed on the BD Accuri C6 flow cytometer. Data were analysed with FlowJo v10 (Becton Dickinson, Wokingham, UK). 1.5 to 2.5 x 105 PBMCs were incubated in pre-coated Fluorospot plates (Human IFNγ FLUOROSPOT (Mabtech AB, Nacka Strand, Sweden)) in triplicate with peptide mixes specific for Spike, Nucleocapsid and Membrane proteins of SARS-CoV-2 (final peptide concentration 1µg/ml/peptide, Miltenyi Biotech) and an unstimulated and positive control mix (containing anti-CD3 (Mabtech AB), Staphylococcus Enterotoxin B (SEB), Phytohaemagglutinin (PHA) (all Sigma Aldrich)) at 37ºC in a humidified CO2 atmosphere for 48 hours. The cells and medium were decanted from the plate and the assay developed following the manufacturer’s instructions. Developed plates were read using an AID iSpot reader (Oxford Biosystems, Oxford, UK) and counted using AID EliSpot v7 software (Autoimmun Diagnostika GmbH, Strasberg, Germany). All data were then corrected for background cytokine production and expressed as SFU/Million CD3 T cells.
SARS-CoV-2 serology
Quantification of Spike SARS-CoV-2 specific antibodies was performed by ELISA as described by Xiong X et al (Xiong et al., 2020). Briefly, serum samples collected at time of enrolment in the study and at the 4-8 week follow-up visit were first screened for positivity and then antibody titres were determined by an end-point analysis. AUC values were calculated in R (3.6.3) using the flux (0.3-0) package. Kruskal–Wallis test was used to calculate p-values among the different disease severities.
Whole blood bulk RNA-Seq
Whole blood RNA was extracted from PAXgene Blood RNA tubes (BD Biosciences) of 188 COVID-19 patients at up to 2 time points and 42 healthy volunteers. RNA-Sequencing libraries were generated using the SMARTer® Stranded Total RNA-Seq v2 - Pico Input Mammalian kit (Takara) using 10ng RNA as input following the manufacturer’s protocol. Libraries were pooled together (n = 96) and sequenced using 75bp paired-end chemistry across 4 lanes of a Hiseq4000 instrument (Illumina) to achieve 10 million reads per sample. Read quality was assessed using FastQC v.0.11.8 (Babraham Bioinformatics, UK), and SMARTer adaptors trimmed and residual rRNA reads depleted in silico using Trim_galore v.0.6.4 (Babraham Bioinformatics, UK) and BBSplit (BBMap v.38.67(BBMap - Bushnell B. - sourceforge.net/projects/bbmap/)), respectively. Alignment was performed using HISAT2 v.2.1.0 (Kim et al., 2019) against the GRCh38 genome achieving a greater than 95% alignment rate. Count matrices were generated using featureCounts (Rsubreads package - (Liao et al., 2019) and stored as a DGEList object (EdgeR package (Robinson et al., 2010) for further analysis.
All downstream data handling was performed in R (R Core Team, 2015). Counts were filtered using filterByExpr (EdgeR) with a gene count threshold of 10 CPM and the minimum number of samples set at the size of the smallest disease group. Library counts were normalised using calcNormFactors (EdgeR) using the method ‘weighted trimmed mean of M-values’. The function ‘voom’ (Law et al., 2014) was applied to the data to estimate the mean-variance relationship, allowing adjustment for heteroscedasticity.
Statistics
All statistical analyses were conducted using custom scripts in R (R Core Team, 2015). Absolute cell counts (cells/uL) were offset by +1 to allow subsequent log2 transformation of zero counts. Where shown, time measures represent time from symptom onset (for severity groups B, C, D and E) or first positive COVID-19 swab (group A). Unless otherwise specified, longitudinally collected data was grouped by bins of 7 or 12 days. Pairwise statistical comparison of absolute cell counts, CRP or serum measures between individuals in a given severity group at a given time bin and HCs, or between severity groups, was conducted by Wilcoxon test unless otherwise specified. For analyses involving repeated measures, false discovery rate corrected (Benjamini & Hochberg) p value were reported. For individuals sampled more than once within a given time bin, data from the earliest blood collection was used.
Cell subset deconvolution of the whole blood RNA-Seq dataset was performed using pathway-level information extractor (PLIER) (http://gobie.csb.pitt.edu/PLIER). Latent factors were generated by leveraging off pre-existing knowledge of cell specific pathways.To better understand the relationship between gene expression and clinical severity, weighted gene co-expression network analysis was carried out using the WGCNA package (Langfelder and Horvath, 2008) in R. Briefly, a signed adjacency matrix was generated and a soft thresholding power was chosen to impose approximate scale-free topology. Modules were identified from the resulting topological overlap matrix with a specified minimum module size of n = 30. Modules were summarized using singular value decomposition, and the resulting module eigengene correlated with clinical traits. Significance of the correlation between a given clinical trait and a modular eigengene was assessed using linear regression with Bonferroni adjustment to correct for multiple testing. Modules were annotated using Enrichr (Chen et al., 2013). Longitudinal mixed modelling of gene module changes over time (yij) was conducted using the nlme package in R (Pinheiro J et al., 2020), including time (tij) with a quadratic trend and disease severity category or unsupervised cluster ids (Xj) as fixed effects, and sampled individuals as random effects (uj):
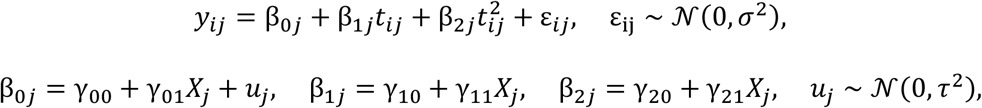 i.e., using the lme formula:
i.e., using the lme formula:
 Gene set enrichment analysis (GSEA) (Subramanian et al., 2005) was used to identify biological pathways enriched in COVID-19 severity groups relative to healthy controls. Briefly, a list of ranked genes, determined by Signal-To-Noise ratio was generated. An enrichment score was calculated, determined by how often genes from the geneset of interest appeared at the top or the bottom of the pre-ranked set of genes with the enrichment score representing the maximum deviation from zero. To assess statistical significance, an empirical phenotype-based permutation test was run, where a collection of enrichment scores was generated from the random assignment of phenotype to samples and used to generate a null distribution. To account for multiple testing, an FDR rate q < 0.20 was deemed significant. A leading edge analysis was performed to determine the genes contributing the most to the enrichment of a given pathway and was subsequently illustrated in a heatmap. HALLMARK gene sets from the Molecular Signatures Database (http://www.broadinstitute.org/gsea/msigdb) were used in analysis.
Gene set enrichment analysis (GSEA) (Subramanian et al., 2005) was used to identify biological pathways enriched in COVID-19 severity groups relative to healthy controls. Briefly, a list of ranked genes, determined by Signal-To-Noise ratio was generated. An enrichment score was calculated, determined by how often genes from the geneset of interest appeared at the top or the bottom of the pre-ranked set of genes with the enrichment score representing the maximum deviation from zero. To assess statistical significance, an empirical phenotype-based permutation test was run, where a collection of enrichment scores was generated from the random assignment of phenotype to samples and used to generate a null distribution. To account for multiple testing, an FDR rate q < 0.20 was deemed significant. A leading edge analysis was performed to determine the genes contributing the most to the enrichment of a given pathway and was subsequently illustrated in a heatmap. HALLMARK gene sets from the Molecular Signatures Database (http://www.broadinstitute.org/gsea/msigdb) were used in analysis.
Principal component analysis (PCA) of centred and scaled absolute counts for 24 major cell types was conducted using the pca()function from the package mixOmics (Rohart et al., 2017). Unsupervised clustering of log2 transformed absolute cell counts, normalised to the median of the corresponding control population, was conducted using the heatmap.2() function from the package gplots (Gregory R. Warnes et al., 2020), with a Euclidean distance function applied to both rows and columns of the data matrix and hierarchical clustering computed using the ward.D method. Partial least squares discriminant analysis (PLS-DA) was conducted using the plsda() function from the package mixOmics (Rohart et al., 2017), a supervised method of sample discrimination whereby sample clustering is informed by group membership (here patient clusters 1 and 2). The classification performance of the PLS-DA model was determined using the perf() function via 10 iterations of 5-fold cross-validation, with two components deemed sufficient to minimise the balanced error rate of prediction. Variable selection on components 1 and 2 was conducted using the tune() function, with 13 cell types selected as those most strongy contributing to discrimination of patient clusters. An AUROC curve showing the performance of a predictive model based on these 13 cell types was generated using the auroc() function. To assess whether clinical severity was reflected on a transcriptional level in an unsupervised fashion, K-means clustering was utilised on normalised whole blood RNASeq gene expression counts. Heat maps were created using the ComplexHeatmap package (Gu et al., 2016), with data scaled and centred prior to visualisation.
Cellular recovery rates over 60 days were calculated for each cell type in patients from groups C, D and E, split into those with persistently elevated (>10mg/L) or resolving CRP (falling below 10mg/L by final bleed), over 60 and 40 days respectively. Using a 12 day sliding window with single day increments, the ‘window of recovery’ for each given cell population was defined as the window in which absolute cell counts for COVID-19 samples no longer differed significantly from controls when assessed by Wilcoxon test, and remained as such for the subsequent 7 windows, and 80% of all windows remaining. Recovery rate was taken as the log2 normalised ratio of test and control absolute counts for patient samples collected within the first time window (0-12 days), subtracted from the equivalent value calculated within the window of recovery, divided by the upper day boundary of the recovery window.
The relationships between immunophenotyping and transcriptional data in the form of gene expression modules were assessed using Pearson’s correlation (Hmisc package) and visualized with corrplot.
Acknowledgements
We thank all the patients and Health Care Workers who consented to take part in this study. We are grateful for the generous support of CVC Capital Partners, the Evelyn Trust (20/75), UKRI COVID
Immunology Consortium, Addenbrooke’s Charitable Trust (12/20A) and the NIHR Cambridge Biomedical Research Centre for their financial support. K.G.C.S. is the recipient of a Wellcome Investigator Award (200871/Z/16/Z); M.P.W. is the recipient of Wellcome Senior Clinical Research Fellowship (108070/Z/15/Z); C.H. was funded by a Wellcome COVID-19 Rapid Response DCF and the Fondation Botnar; N.M. was funded by the MRC (CSF MR/P008801/1) and NHSBT (WPA15-02); I.G.G. is a Wellcome Senior Fellow and was supported by funding from the Wellcome (Ref: 207498/Z/17/Z). We would like to thank the NIHR Cambridge Biomedical Research Centre Cell Phenotyping Hub and the CRUK Cambridge Institute flow cytometry core facility for their support with flow and mass cytometry.
Footnotes
The CITIID-NIHR COVID-19 BioResource Collaboration
Principal Investigators Stephen Baker, John Bradley, Gordon Dougan, Ian Goodfellow, Ravi Gupta, Nathalie Kingston, Paul J. Lehner, Paul A. Lyons, Nicholas J. Matheson, Willem H. Owehand, Caroline Saunders, Kenneth G.C. Smith, Charlotte Summers, James E. D. Thaventhiran, Mark Toshner, Michael P. Weekes and Christoph Hess
CRF and Volunteer Research Nurses Ashlea Bucke, Jo Calder, Laura Canna, Jason Domingo, Anne Elmer, Stewart Fuller, Julie Harris, Sarah Hewitt, Jane Kennet, Sherly Jose, Jenny Kourampa, Anne Meadows, Criona O’Brien, Jane Price, Cherry Publico, Rebecca Rastall, Carla Ribeiro, Jane Rowlands, Valentina Ruffolo and Hugo Tordesillas
Sample Logistics Ben Bullman, Benjamin J. Dunmore, Stuart Fawke, Stefan Gräf, Josh Hodgson, Christopher Huang, Kelvin Hunter, Emma Jones, Ekaterina Legchenko, Cecilia Matara, Jennifer Martin, Federica Mescia, Ciara O’Donnell, Linda Pointon, Nicole Pond, Joy Shih, Rachel Sutcliffe, Tobias Tilly, Carmen Treacy, Zhen Tong, Jennifer Wood and Marta Wylot
Sample Processing and Data Acquisition Laura Bergamaschi, Ariana Betancourt, Georgie Bower, Chiara Cossetti, Aloka De Sa, Madeline Epping, Stuart Fawke, Nick Gleadall, Richard Grenfell, Andrew Hinch, Oisin Huhn, Sarah Jackson, Isobel Jarvis, Daniel Lewis, Joe Marsden, Francesca Nice, Georgina Okecha, Ommar Omarjee, Marianne Perera, Nathan Richoz, Veronika Romashova, Natalia Savinykh Yarkoni, Rahul Sharma, Luca Stefanucci, Jonathan Stephens, Mateusz Strezlecki and Lori Turner
Clinical Data Collection Eckart M.D.D. De Bie, Katherine Bunclark, Masa Josipovic, Michael Mackay, Federica Mescia, Alice Michael, Sabrina Rossi, Mayurun Selvan, Sarah Spencer and Cissy Yong
Royal Papworth Hospital ICU Ali Ansaripour, Alice Michael, Lucy Mwaura, Caroline Patterson and Gary Polwarth
Addenbrooke’s Hospital ICU Petra Polgarova and Giovanni di Stefano
Cambridge and Peterborough Foundation Trust Codie Fahey and Rachel Michel
ANPC and Centre for Molecular Medicine and Innovative Therapeutics Sze-How Bong, Jerome D. Coudert and Elaine Holmes
NIHR BioResource John Allison, Helen Butcher, Daniela Caputo, Debbie Clapham-Riley, Eleanor Dewhurst, Anita Furlong, Barbara Graves, Jennifer Gray, Tasmin Ivers, Mary Kasanicki, Emma Le Gresley, Rachel Linger, Sarah Meloy, Francesca Muldoon, Nigel Ovington, Sofia Papadia, Isabel Phelan, Hannah Stark, Kathleen E Stirrups, Paul Townsend, Neil Walker and Jennifer Webster
References




Subject Area
Reviews and Context
1
Comment
0
TRIP Peer Reviews
0
Community Reviews
0
Automated Services
24
Blogs/Media
Author Videos