
Abstract
The interplay between the virus, infected cells and the immune responses to SARS-CoV-2 is still under debate. Extending the basic model of viral dynamics we propose here a formal approach to describe the neutralizing versus non-neutralizing scenarios and compare with the possible effects of antibody-dependent enhancement (ADE). The theoretical model is consistent with data available from the literature and conclusions show that, while both non-neutralizing scenarios and ADE give rise to similar final virus clearance, the non-neutralizing antibodies can induce permanent high levels of antibody production with documented unfavorable impact on the disease progression and outcome. We also discuss the implications on secondary infections.
1 Background
SARS-CoV-2 is a new virus from the coronavirus family, responsible for the ongoing COVID-19 pandemic. To date, there are more than 35 million cases and over a million deaths worldwide [1]. SARS-CoV-2 is the third betacoronavirus to severely infect humans appearing in the last 20 years, after SARS-CoV-1 and MERS-CoV. This motivates a growing need for efficient drugs and/or vaccines, not only for the time being but also in anticipation of a future coronavirus resurgence.
However, initial promising successes of antiviral treatments raised also the possibility of negative side-effects. On the vaccine front, an auto-immune disease (leading to temporary suspension of clinical trials) appeared during the AstraZeneca vaccine trial (Sept 9th 2020); altogether this context demonstrated the importance of understanding qualitatively and quantitatively the immune response to primary infection and also to challenges (vaccines belong to both categories). In particular, relevant mathematical models of the immune dynamics can be of interest to understand and predict the complicated behavior often observed.
We focus here on adaptive humoral immunity (the antibody-mediated immunity) and refer to future works for an extension to the cellular and/or innate immune system. In particular, our goal is to investigate the nature of the response to secondary (challenge) infection using a viral dynamics model.
1.1 Available evidence detailing the antibody response
For clinical reasons and also for the understanding of those studying vaccines, antibody responses are of paramount importance; SARS-CoV-2 specific antibodies are usually detected during the second week after illness onset (see [2, 3]) and remain active thereafter for an unknown time span (see however [4] for recent information). Antibody responses are mainly directed against the RBD-spike and nucleocapsid proteins. However, the neutralization capacities of these specific antibodies is still under discussion, especially since non-neutralizing antibodies can enhance infection through a process called antibody-dependent enhancement (hereafter abbreviated ‘ADE’) [5, 6, 7, 8]. This has been recently emphasized in the set up of clinical trials (see for example [9]), in a general discussion of the prospects of vaccination [10] and in a perspective accounting for the present situation in terms of SARS-CoV-2 vaccines, therapies and immunity [11, 12]
The present academic interpretation of the ADE is that it occurs through virus-antibody immunocomplexes that facilitate virus internalization in host cells that do not express virus receptor but Fc receptors. ADE is induced when the antibody-virus stoichiometry is below the threshold for neutralization, [5, 6]. As a consequence, neutralizing antibodies may enhance infection when their concentrations fall below a key occupancy threshold, and some poorly neutralizing antibodies may strongly increase infection over a wide dose–response range. ADE has been demonstrated in vitro for many viral infections, including that triggered by SARS-CoV-1 which was reported to infect in vitro human macrophages (see [7]) and human B cell lines via an ADE pathway, (see [8]). Moreover, Qidi Wang et al. reported that a specific spike protein epitope elicited antibodies which could enhance infection via ADE, while other epitopes induced neutralizing antibodies in non-human primates. Furthermore, the authors showed that a SARS-CoV-1 inactivated vaccine could induce ADE and lung pathology in experimental rhesus monkeys [13]. In contrast, Martial Jaume and co-authors showed that vaccine candidates which mediated in vitro ADE infection could still be neutralizing and protective in vivo on rhesus macaques, [8]. Moreover, in most cases, ADE infected cells do not support viral replication, [7, 8]. Instead, ADE may trigger cell apoptosis and promote tissue inflammation and injury with the release of pro-inflammatory cytokines from infected cells, [6, 14]. As a result, whether ADE actually happens in SARS-CoV-1 infected humans and is a factor of disease severity is still a debated research subject since no in vivo human evidence has been demonstrated yet (but this statement is very time-dependent given the present intense research on SARS-CoV-2). Note however, that SARS-CoV-1 infected patients who developed a higher and earlier antibody response were associated with worse clinical outcome. An early antibody response may be weakly neutralizing compared to a later one. As a consequence, a high concentration of those antibodies could lead to ADE and enhancement of infection.
The question of ADE and the link between antibody dynamics and disease evolution is still unclear for COVID-19. J. Zhao et al. reported a strong positive correlation between disease severity and high antibody titers two weeks after illness onset. The antibody level is considered as a risk factor for severe evolution, independently from age, gender and comorbidities [15]. In another study, Wenting Tan et al also came to the same conclusion: higher titers of anti-N IgM and anti-N IgG are observed for severe patients [16]. Finally, Baoqing Sun et al observed that severe patients had higher levels N-IgG than S-IgG after the symptoms onset. However, according to the authors, whether N-specific antibodies can block virus infection is still open to question [17]. The secretion of a high level of non-neutralizing antibody supports the hypothesis of ADE for COVID-19 which can partially explain some clinical complications. In contrast, Mehul S. Suthar et al concluded that the appearance of high titer neutralizing antibody responses early after the infection was promising and may offer some degree of protection against re-infection [18]. This result seems to be confirmed in a recent study in which SARS-CoV-2 infection induced protective immunity against re-exposure in nonhuman primates. However, rhesus macaques do not develop severe clinical complications as reported in human patients, suggesting that if rhesus macaques produce neutralizing antibodies, transposition of this observation to humans is still to be investigated [19]. Finally, a recent study on a recovered cohort of COVID-19 patients showed that elderly patients had significantly higher levels of antibodies than younger patients. However, severe and critical patients were excluded from the study because they received passive antibody treatment before sample collection. As a result, the authors could not directly evaluate the effect of antibodies on virus clearance or disease progression in COVID-19 patients [20]. This suggests that if elderly patients tend to develop higher titers of antibodies, those may not be systematically associated with worse clinical evolution. What should rather be answered is whether disease severity is systematically associated with high antibody levels.
On the other hand, the vaccine community is increasingly aware of this need (see discussion on ADE in [11, 10, 12]) and studies along these lines are required.
Another motivation comes from the fact that the adaptive immune system response starts in about a week; on the other hand in many mild forms infection is resolved in around a week while on the contrary severe forms may at first start as mild and only then become severe; a simplistic view may indicate that the innate immune response is very efficient while the adaptive immune system response may be detrimental. In this case, everyone with a mild first infection (i.e., mostly dealt with by the innate immune system) will, upon re-infection, see a adaptive immune rising faster (once the memory is in place, its response is faster than the innate immune response) and thus the detrimental effects could be visible for people previously having experienced mild forms, e.g., low age class individuals.
1.2 Evidence on re-infection
The possible unfavorable outcomes of a secondary infection (challenge) following a primary SARS-CoV-2 infection were described in various situations (see for example ¡10.1093/cid/ciaa1436¿), but an increasing body of evidence highlights the Kawasaki-like syndrome as a possible negative outcome, see [21, 22, 23, 24, 25].
An italian study [23] indicates that the immune response to SARS-Cov-2 is responsible for the appearance of a pediatric Kawasaki-like syndrome (Kawasaki-like disease or Multisystem Inflammatory Syndrome in Children MIS-C in the US). In this study, 8 to 10 children have been tested positive to IgG, IgM or both (the infection to SARS-CoV-2 preceded the development of the syndrome) and 2 only in PCR (the infection was simultaneous). SARS-CoV-2 infected children who developed the Kawasaki-like syndrome (KLS) were on average older and more severely hit than other children victims of the classical Kawasaki syndrome.
The same phenomena has been observed in the US and UK [21, 24, 25]. Academic studies begin to investivage the interplay between COVID-19 and the MIS-C [26].
The causes of the development of the Kawasaki disease are still unknown. The best accepted hypothesis is that of an abnormal immune response that occurs as a result of the infection provoked by one of several pathogenic agents for the genetically susceptible patients. The triggering pathogens have not yet been identified.
To account for the peaks of the KLS cases following an infection with SARS-CoV-2, two hypothesis may be formulated: the antibodies produced by the children can induce the initiation of an autoimmune disease and syndromes similar to the Kawasaki syndrome. The second hypothesis is an ADE-type mechanism.
1.3 Background summary
To summarize, the question of antibody protection and immune system reaction to a virus is still largely under debate. To progress in this discussion, we investigated here the role of ADE in the pathogenesis of COVID-19 in a primary and secondary infection. We propose a mathematical model of the immune response and virus dynamics that includes the possibility of non-neutralizing antibodies and / or ADE.
2 Methods
2.1 Mathematical model
We present below the viral and immune response model. The viral-host interaction (excluding the immune response) is called the basic model of virus dynamics. It has been extensively validated both theoretically and experimentally, see [27, eq (3.1) page 18], [28, eqns. (2.3)-(2.4) page 26] and references therein. See also [29, 30, 31] for general overviews of mathematical immunology.
The model involves several classes: that of the target cells, denoted T, the infected cells, denoted I, the free virus denoted V and the antibodies denoted A. The model is illustrated in figure 1.
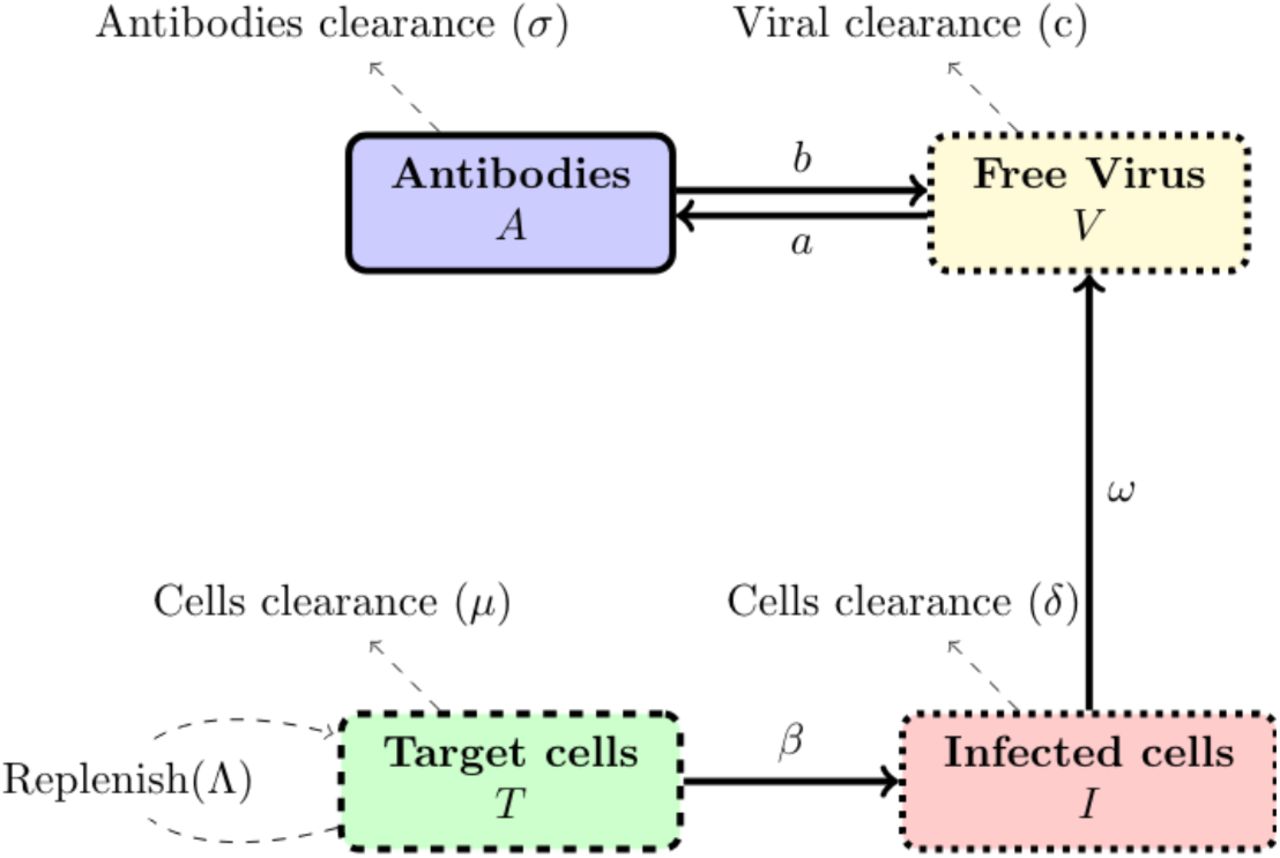
Graphical illustration of the flow in the model (1)-(5).
Target cells T, which in our case are the epithelial cells with ACE2 receptors located, for instance in the respiratory tracts including lungs, nasal and trachea/bronchial tissues, are produced at a rate Λ and die at rate µ. The parameters Λ and µ define tissue dynamics in the absence of infection, see also section A.1. When these susceptible cells meet free virus particles V, they become infected at a rate β0. Furthermore, target cells can also become infected via ADE if virus entry is mediated by antibodies. The parameter β1 represents the rate of ADE infection route which is the result of a three-species interaction: T, A and V.
Infected (initially target) cells, denoted I, die at a rate δ. Note that this death rate will often be larger than the death rate of uninfected cells because viruses cause cell damage and cell death, [28, 27]. Infected cells produce new virus particles at a rate ω, and the free virus particles which have been released from infected cells decay at a rate c called the clearance rate.
Free virions are neutralized by antibodies A, which can block virus entry into cells but also facilitate phagocytosis, at a rate b. Finally, the antibodies can be stimulated by the free virus with a production rate a while it declines at a rate of σ (see for instance [28, eq. (9.4) p.126]). The complete model reads (all constants are positive):




 Several hypotheses in this model need to be further documented. The first one in that all infected cells including ADE infected cells support viral replication and can produce virus. However, to date, it is still unclear whether ADE infected cells can support viral replication in vivo, [7], [8]. Here we choose not to distinguish between virus productive and non productive infected cells to keep the model simple. For the same reason, we do not discriminate between neutralizing and non neutralizing antibodies but consider both as members of the same class, the antibodies neutralizing capacity will therefore be the average of the neutralizing and non-neutralizing species and the average is described by the parameter b; on the other hand the ADE magnitude will be monitored by parameter β1. These parameters are the most important part of the immune response and the object of our study.
Several hypotheses in this model need to be further documented. The first one in that all infected cells including ADE infected cells support viral replication and can produce virus. However, to date, it is still unclear whether ADE infected cells can support viral replication in vivo, [7], [8]. Here we choose not to distinguish between virus productive and non productive infected cells to keep the model simple. For the same reason, we do not discriminate between neutralizing and non neutralizing antibodies but consider both as members of the same class, the antibodies neutralizing capacity will therefore be the average of the neutralizing and non-neutralizing species and the average is described by the parameter b; on the other hand the ADE magnitude will be monitored by parameter β1. These parameters are the most important part of the immune response and the object of our study.
3 Results
3.1 Theoretical results
We refer the reader to the Appendix C for the rigorous statements concerning the theoretical properties of the model (1)-(5). We analyzed the equilibria of the model but the main conclusion is that stochastic events prevent the stable equilibrium state to be reached in practice, cf. section A.5. The parameters b and β1 are shown to be the most important for the viral-host-antibody dynamics.
3.2 Empirical results
Takins into account the available data from the literature and the methodology in section B we used as baseline the following parameters:
We focus on the following scenario :
Step 1: the patient undergoes a first infection of SARS-CoV-2 without ADE (β1 = 0).
Step 2: The infection is controlled and the patients is cured few weeks after infection.
Step 3: A few months later, there is a challenge from a slightly different new coronavirus which triggers ADE (β1 > 0).
The numerical simulation for the first infection corresponding to parameters in table 1 is shown in figure 2.
Baseline parameters use in numerical simulations of the model (1)-(4)
Baseline parameters and references use in numerical simulations of the model (1)-(4)

Numerical simulation of the first infection without ADE for model (1)-(4) and parameters in table 1.
There is a 10% fall of target cells which either become infected or naturally die. The viral load peaks around day 5 after symptoms onset at 106 copies/ml. While SARS-CoV-1 viral load, as MERS-CoV, peaked around 10 days after symptoms onset, most studies agree that SARS-CoV-2 viral load peaks sooner, around day 5, [32],[33]. Concerning antibodies, they increase sharply until week 2 then slower until a month after infection and start to decrease within 2-3 months [34], [35]. Qualitative agreement is observed with clinically observed variations variations of viral load and antibodies concentration depicted in figure 3 (see references in the figure).
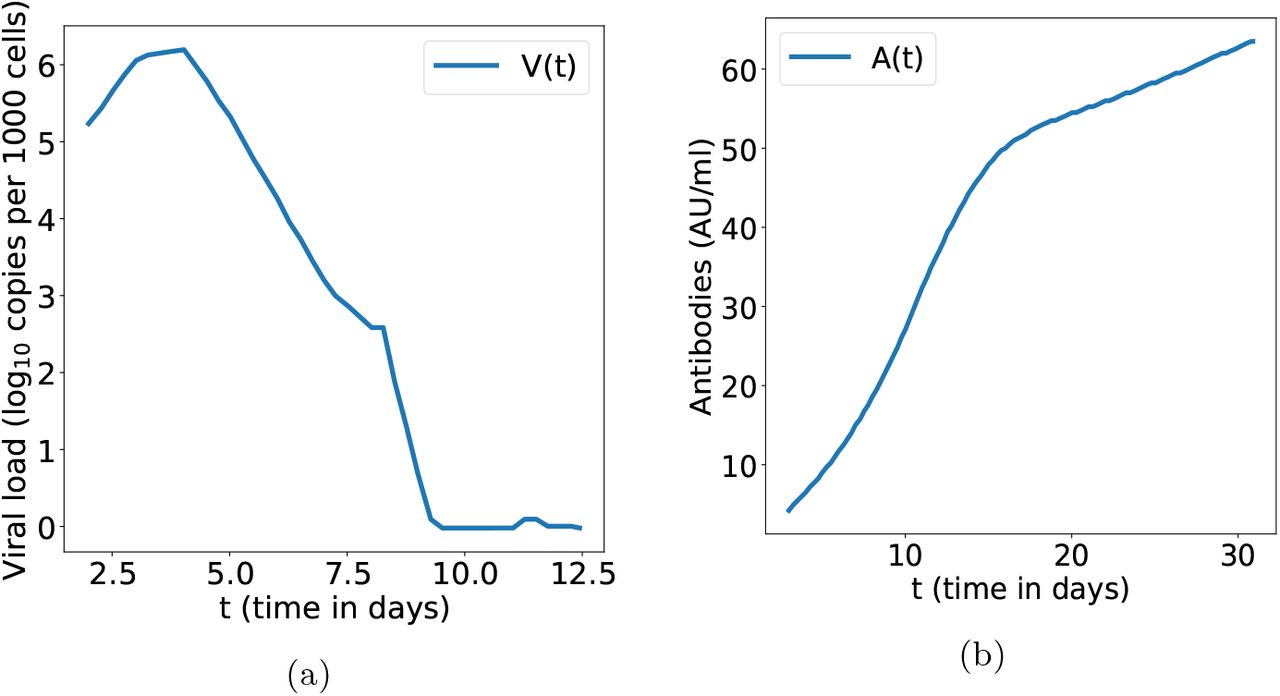
3a : Clinically observed typical variation of SARS-CoV-2 viral load in nasopharyngeal swab normalised using cell quantification. Data taken from [32, figure 3 page 703]. 3b : Typical time variations for IgG. Data taken from [36, figure 2 page 1085].

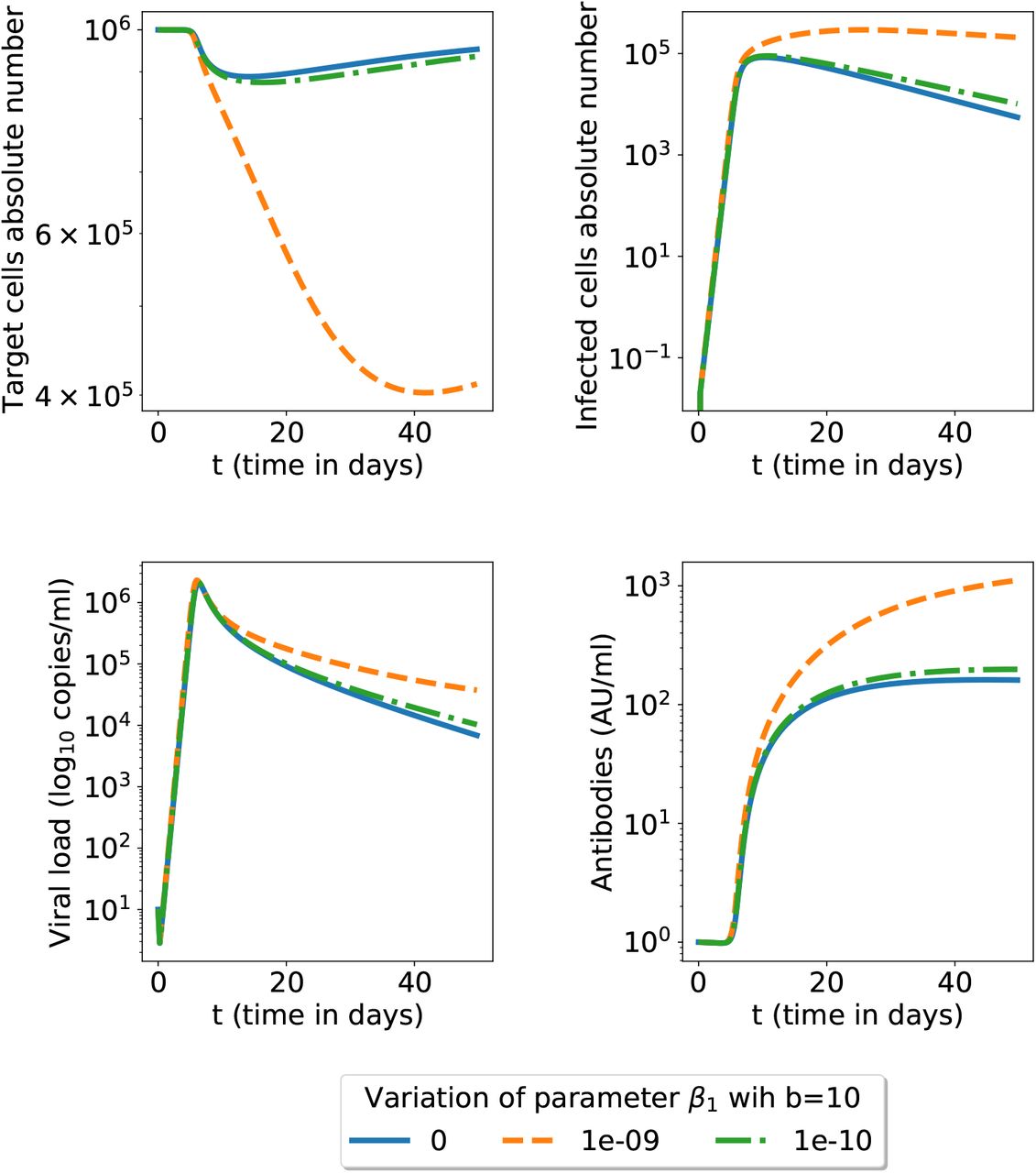
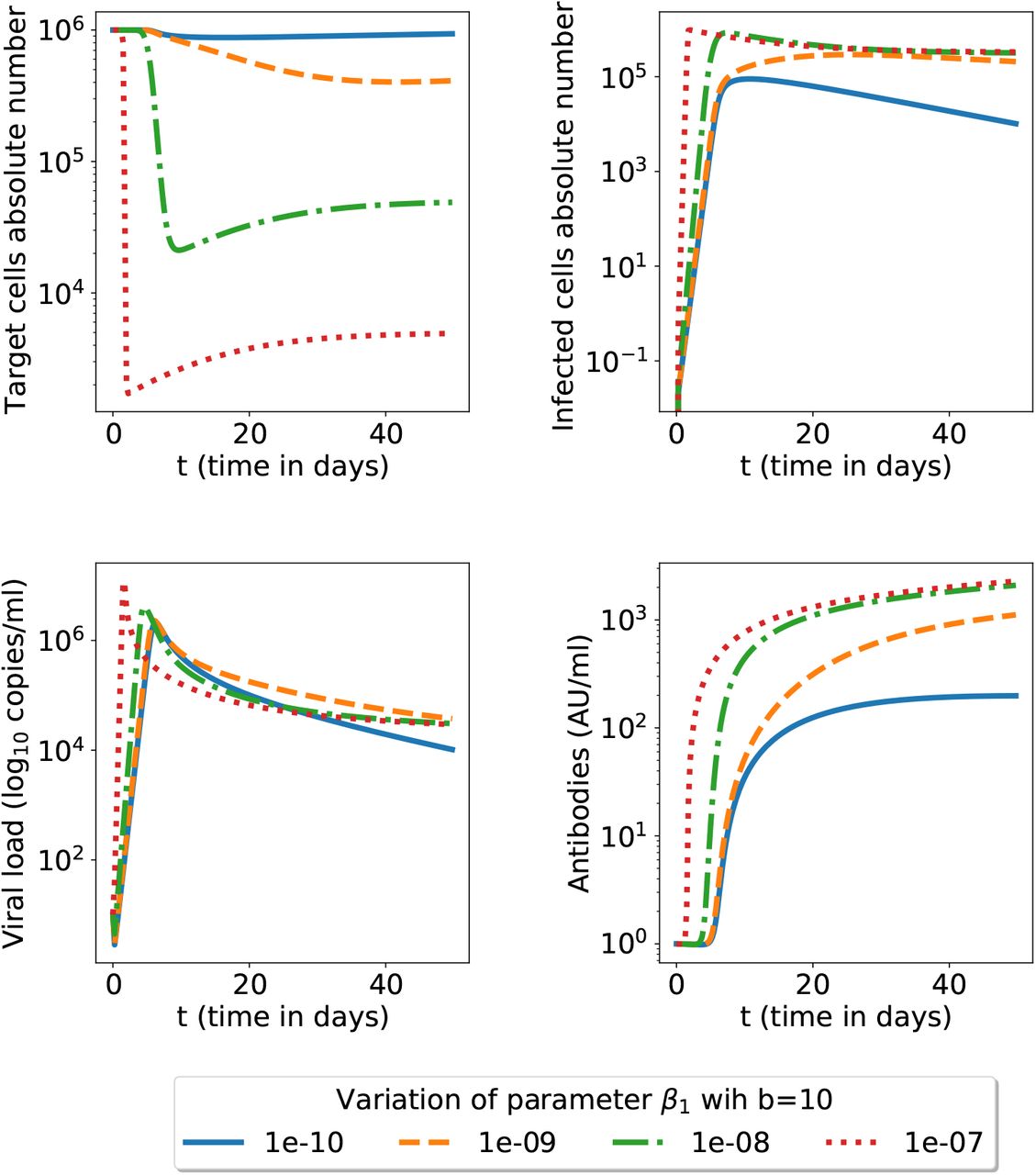
The effect of the ADE parameter β1 (the model (1)-(4)). The secondary infection has fixed neutralizing antibodies capacity b but we investigate several β1 (ADE) parameter values; for all other parameters we use the nominal values given in table 1.

The effect of the neutralizing b parameter. The secondary infection has lower neutralizing antibodies capacity b = 3.16. We fixed ADE parameter β1 and considered all other parameters of the model (1)-(4) at their nominal values in table 1.
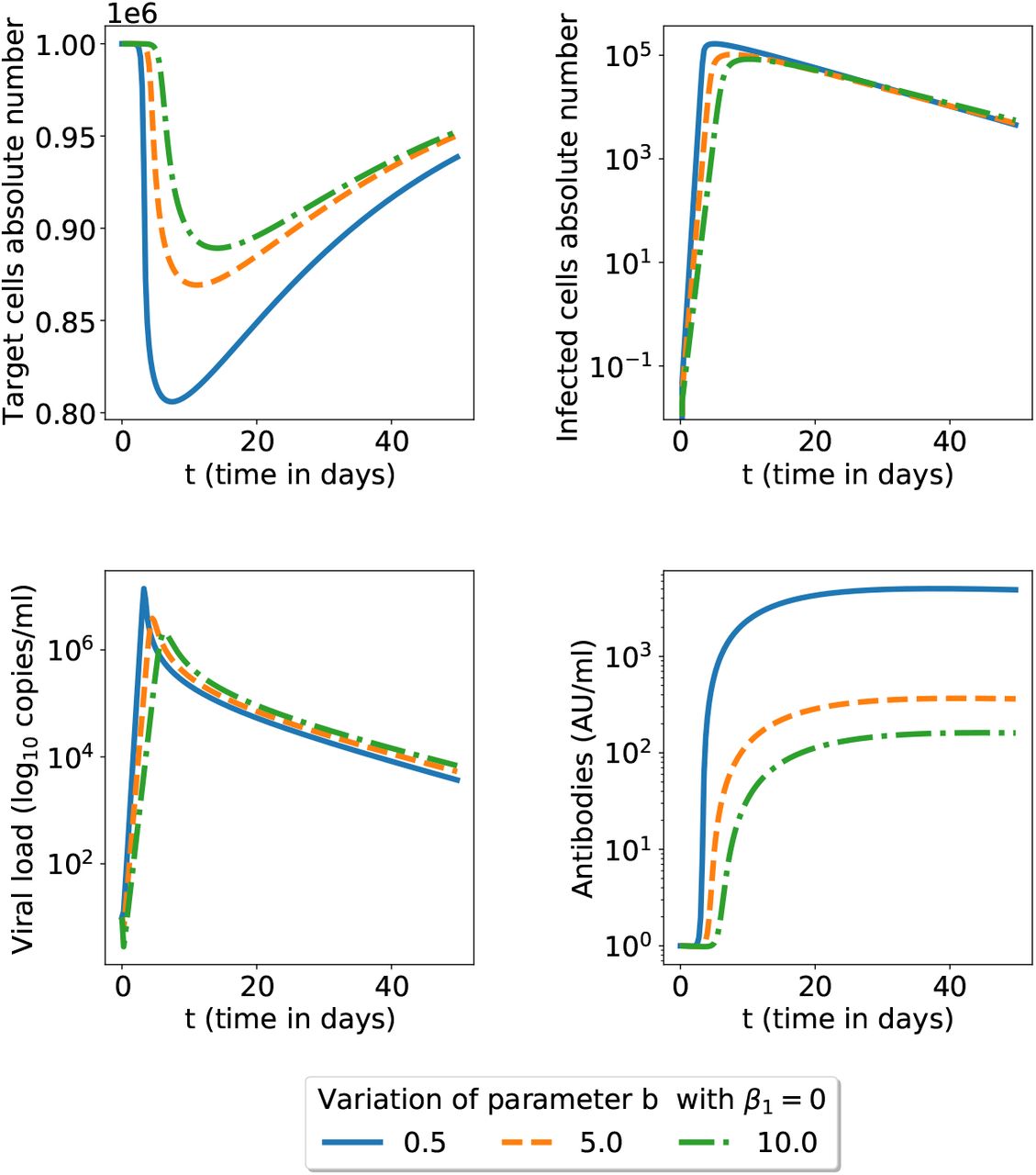
Initial infection for three possible values of the b parameter; the model is that in equations (1)-(4) and all other parameters have the nominal values in the table 1. We show here a zoom for the initial 50 days (see figure 7 for a longer time span).
The equilibrium state (19) when β1 = 0 (no ADE present) is reached after 4 years for all variables in figure 2. However, viral load and infected cells reach a minimum close to 0 within 6 month post-infection before increasing and oscillating toward equilibrium state (19) (simulations not shown here). Therefore, if the virus load is null close to the minimum, all other variables decrease towards 0 and the infection has vanished. The equilibrium state (19) is stable but not reached in practice as the patient is cured. When there is no ADE, decreasing the neutralizing capacities of antibodies leads on the one hand to a higher viral load peak but on the other hand to higher antibodies concentrations. The less neutralizing the more abundant antibodies are to compensate so that the infection is always solved (see figure 7).
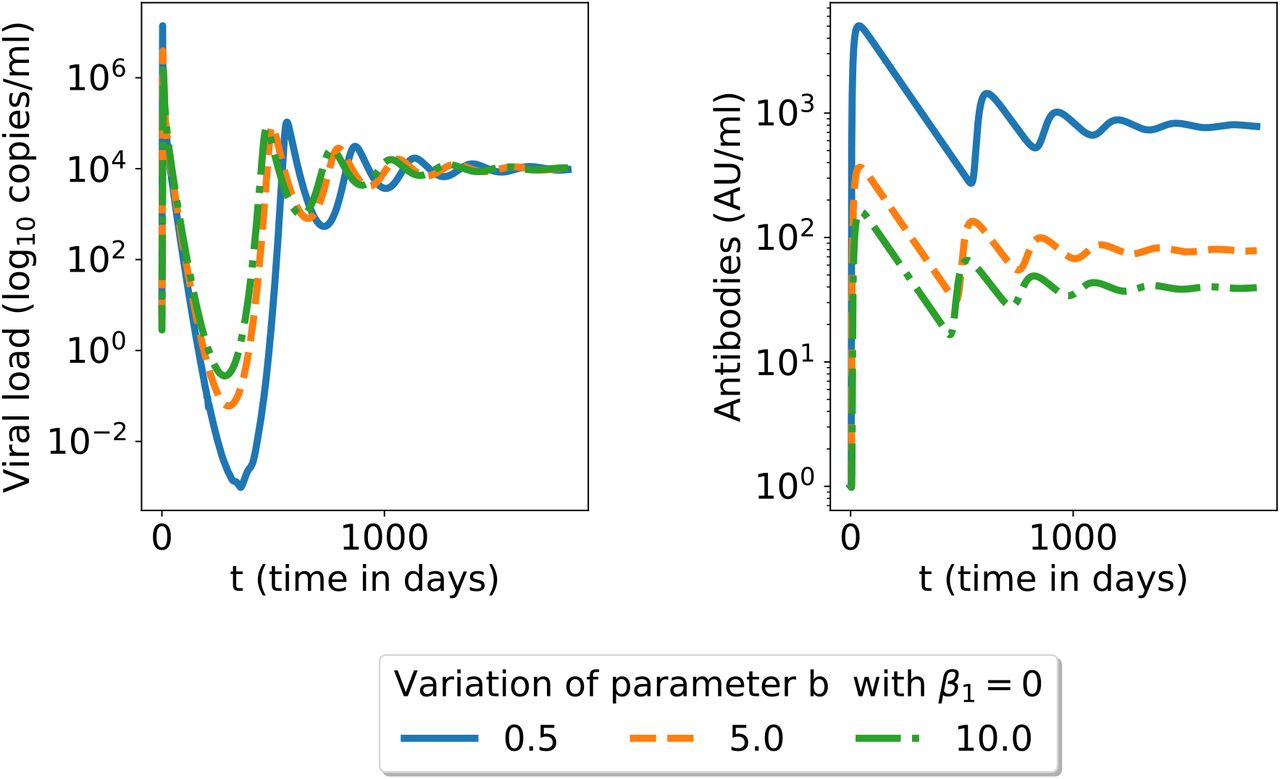
Simulation in 6 for a longer time span.
Next we investigated in figure 4 the possibility of the ADE mechanism present (β1 > 0), for a range of possible parameter β1 values. We plot all variables upon challenge with the same neutralizing capacity for antibodies. ADE can be triggered by several mechanisms, such as non-neutralizing antibodies or sub-optimal concentration of neutralizing antibodies, which, for simplicity, are not distinguished in this model. A higher ADE parameter leads to more destroyed target cells, more infected cells, more viral load and more antibodies. However the antibodies concentration is restricted by an upper limit. There is a threshold when increasing β1 does not increase significantly the antibody population (see figure 9). Therefore a higher β1 ADE parameter cannot be compensated by more antibodies as a lower neutralizing capacity was already present for the first infection. For example, unlike β1 = 10−10 and β1 = 0, if β1 = 10−9 the viral load directly stabilises to its equilibrium state (21), without reaching a minimum close to 0 while oscillating (simulation not shown here). In this case, the infection wins (leading to respiratory function disruption and possibly patient death).

Secondary infection with ADE mechanism present for several values of the β1 parameter; the model is that in equations (1)-(4) and all other parameters have the nominal values in the table 1. We show here a zoom for the initial 50 days (see figure 9 for a longer time span).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simulation in 8 for a longer time span
Next we performed the contrary analysis: we used, for the secondary infection, two possible values (b = 3.16 and b = 10) of the parameter b (i.e., antibodies neutralizing capacity) while fixing β1. In this case the viral load does not significantly differ between the values b = 3.16 and b = 10, but antibodies and destroyed target cells change more dramatically and are significantly higher during the first weeks of the disease, (see figure 5). For b = 3.16, the viral load still oscillates but do not reach small values, its minimum being 3 × 102 copies/ml, before increasing and stabilizing towards the equilibrium state described in formulas (21).
4 Discussion
We investigated the immune response to SARS-CoV-2 infection and re-infection through a numerical model; the model that can also take into account the possible presence of ADE, either on first infection or to a challenge (secondary or re-infection with a different phenotype, vaccine, etc.).
As to date there is no clear evidence that ADE occurs in COVID-19 severe patients, we assume that ADE only happens upon challenge.
We started from a classic viral-host dynamics ([28],[37]) that we modified by adding parameter β1 to account for a possible ADE mechanism. In order to keep the model at its lowest complexity, we do not distinguish between ADE triggering antibodies and neutralizing antibodies.
We conducted a theoretical study of our system by computing equilibrium states and stability with and without ADE. We showed that stochastic events prevent the stable equilibrium state to be reached in practice and identified parameters b and β1 to be crucial in the precise dynamics of our system.
Then, we calibrated our parameters values in coherence with references from the literature and available clinical data published in recent studies, [32], [35], on viral load and antibodies concentration. We tested our model with our scenario. We saw that without ADE, the antibody lower neutralizing capacity was systematically compensated with higher concentrations of antibody leading to viral clearance. On the other side, adding ADE was not always associated with viral clearance. Simulations and equilibrium analysis showed that antibody concentration had an upper limit which prevented higher ADE to be compensated by an unlimited antibody quantity. Therefore, ADE should be taken in consideration as a serious risk in vaccine development or disease understanding and treatment.
On the other hand, we showed that the results are sensitive to the neutralizing antibody capacity (the b parameter); note that a decrease of this parameter can occur in several situations, for instance due to immune function decay, due to the malfunctioning of the antibody immunodominance mechanism that ends up selecting too many weakly neutralizing antibodies or due to miscalibrated therapeutic interventions. Independent of the cause, such a decrease of the neutralizing capacity is susceptible to imply a substantial deterioration of the outcome.
As any other, our model contains of course several limitations. First, we considered all infected cells to support viral replication, including ADE-infected cells. Concerning SARS-CoV-2, the questions of ADE is still under debate, but for SARS-CoV-1 in vitro ADE evidence suggested abortive viral replication in ADE infected cells. Therefore, if we changed the model (1)-(4) to include this distinction, equilibrium state would change and ADE may be compensated. Similarly, we did not distinguish between former antibodies and novel antibodies secreted upon challenge. This would imply more parameters and change equilibrium levels but without inherently changing variables behaviour. Regarding parameters, we did not have enough exploitable available data to train our model and fit parameters better. Finally, an unique model can hardly capture the extreme variability of COVID-19 clinical outcomes, see [38]; some studies proposed that some of the variability come from genetics, see e.g., [39] where genetic information from roughly 4,000 people from Italy and Spain was correlated to severity of COVID-19. This may lead to a variability of our model parameters in the form of random variables.
The more science will shed light on the full picture of SARS-CoV-2, the more our model can input complex and precise details. In the meantime, the main take-home message is that, with parameters consistent with the available clinical data, the neutralizing capacity and ADE mechanisms may play an important immunological role into the primary and secondary infection outcomes.
A Mathematical details
We describe in an incremental way the mathematical properties of the main model (1)-(5).
A.1 Model without a virus, nor immune response
In absence of any infection the equations for the target cells are (see [28, 27]):
 Since the Jacobian matrix at equilibrium (a 1 × 1 matrix) is the constant −µ therefore the equilibrium is stable, in fact any initial data T (0) will converge to the equilibrium
Since the Jacobian matrix at equilibrium (a 1 × 1 matrix) is the constant −µ therefore the equilibrium is stable, in fact any initial data T (0) will converge to the equilibrium

A.2 Model with virus but no immune response
We employ the basic model of virus dynamics, see [27, eq (3.1) page 18] and also [28, eqns. (2.3)-(2.4) page 26] described by the equations:


 The initial conditions are:
The initial conditions are:
 which express the fact that the initial state for T is the stable equilibrium seen in section A.1, there are initially no infected cells and the initial viral load is strictly positive.
which express the fact that the initial state for T is the stable equilibrium seen in section A.1, there are initially no infected cells and the initial viral load is strictly positive.
It is natural to assume that the decay rate of infected cells is at least as large as the decay rate of healthy cells, i.e.,
 In this model, an infection is only possible if the basic reproduction ratio of the virus in the absence of immune response, defined as (cf. [27, eq. (6.2) page 53])
In this model, an infection is only possible if the basic reproduction ratio of the virus in the absence of immune response, defined as (cf. [27, eq. (6.2) page 53])
 is strictly super-unitary, that is
is strictly super-unitary, that is
 Otherwise, that is if R0 ≤ 1, the initial viral load can only decrease. The model has two critical points (equilibrium candidates):
Otherwise, that is if R0 ≤ 1, the initial viral load can only decrease. The model has two critical points (equilibrium candidates):
-critical point: T = T∗ = Λ/µ, V = I = 0. The Jacobian matrix at equilibrium is  . The eigenvalues of this matrix, under condition (14), are all real but not all negative: one of them is λ1 = −µ but the product of the other two is δc − ωβ0T∗ ≤ 0 thus at least one is positive. Therefore this critical point is not an equilibrium.
. The eigenvalues of this matrix, under condition (14), are all real but not all negative: one of them is λ1 = −µ but the product of the other two is δc − ωβ0T∗ ≤ 0 thus at least one is positive. Therefore this critical point is not an equilibrium.
-critical point called “immunosuppression” state:
 For values in table 1, we obtain : T is = 4.55 × 108, V is = 2.18 × 108 and Iis = 3.28 × 105.
For values in table 1, we obtain : T is = 4.55 × 108, V is = 2.18 × 108 and Iis = 3.28 × 105.
The Jacobian matrix is  ; the characteristic polynomial P (X) = (X +δ)(X +c)(X +µ +β0V is) δc(X +µ) has the following properties: P (−∞) < 0, P (−δ −c) = δcβ0V is > 0, thus it has a real root which is smaller than −δ −c. The product of all roots is δcβ0V is > 0 and the sum of all roots is −δ −c −µ −β0V is < −δ −c, thus the other two roots have negative real part. Therefore the equilibrium is stable.
; the characteristic polynomial P (X) = (X +δ)(X +c)(X +µ +β0V is) δc(X +µ) has the following properties: P (−∞) < 0, P (−δ −c) = δcβ0V is > 0, thus it has a real root which is smaller than −δ −c. The product of all roots is δcβ0V is > 0 and the sum of all roots is −δ −c −µ −β0V is < −δ −c, thus the other two roots have negative real part. Therefore the equilibrium is stable.
It is important to note that the viral load V is is the viral load that the infection will cause in a completely immunodeficient individual. We expect V is to be significantly high, see in section A.3 for details.
A.3 Model: virus and immune response but no enhancement
In this section we consider the model (1)-(4) with no ADE i.e., β(A) = β0 that is β1 = 0. This model is similar to other in the literature (see for instance [28, eq. (2.9) page 29] who consider also the cytotoxic effect of the immune response on the infected cells; however they do not consider virus destruction by antibodies. In particular there virus load is constant. Another similar model is [28, eqns. (8.1)-(8.3)]. With respect to the previous section here the immune response is present. It is triggered by a threshold set at
 It is natural to suppose that the immune response threshold is a very small value and in any case a value smaller than the immunosupression viral load V is in (15). That is we can make the hypothesis that V is > V t i.e.
It is natural to suppose that the immune response threshold is a very small value and in any case a value smaller than the immunosupression viral load V is in (15). That is we can make the hypothesis that V is > V t i.e.
 The Jacobian matrix is:
The Jacobian matrix is:
 With these provisions, one can find analytically the critical points (equilibria candidates):
With these provisions, one can find analytically the critical points (equilibria candidates):
T = T∗ = Λ/µ, V = I = A = 0, which is the high dimensional analog of equilibrium (7). However, unlike in section A.1, this equilibrium is not stable any more (the determinant of the Jacobian matrix is negative when hypothesis (14) is satisfied.
the immunosuppression equilibrium (15) with A = 0; again this equilibrium is not stable any more because the condition (17) implies that the eigenvalue aV is − σ is positive.
the only critical point left is
 Note that the equilibrium value of the antibody level is positive due to condition (17). For values in table 1 we obtain T = 993976, I = 1988, V = 10000, A = 39 and the Jacobian matrix evaluated at this equilibrium is :
Note that the equilibrium value of the antibody level is positive due to condition (17). For values in table 1 we obtain T = 993976, I = 1988, V = 10000, A = 39 and the Jacobian matrix evaluated at this equilibrium is :
 The eigenvalues are −3.98 × 102, −3.27 × 10−2, −3.20 × 10−3 ± 0.024i. All real parts are negative thus equilibrium is stable.
The eigenvalues are −3.98 × 102, −3.27 × 10−2, −3.20 × 10−3 ± 0.024i. All real parts are negative thus equilibrium is stable.
A.4 Full model: virus, immune system and ADE
We consider the model (1)-(4) with β(A) = β0 + β1A (β1 > 0).
The analysis of this dynamics is more involved. There are two equilibria with A = 0 which are the complete analogues of the equilibria seen in previous sections and have no dynamical interest.
The critical point (19) is replaced by two critical points because the antibody level is solution of the following second order equation:
 and
and
 Note however that equation (20) has exactly one positive solution (the product of roots being negative), thus only a critical point is admissible, which is the positive solution of (20) (with respect to the unknown A), while the other values are obtained as in (21).
Note however that equation (20) has exactly one positive solution (the product of roots being negative), thus only a critical point is admissible, which is the positive solution of (20) (with respect to the unknown A), while the other values are obtained as in (21).
The Jacobian matrix is:
 For values in table 1 and β1 = 10−10 we obtain T∗ = 9925061, I∗ = 2472, V ∗ = 10000, A∗ = 49 and the Jacobian matrix evaluated at this equilibrium is :
For values in table 1 and β1 = 10−10 we obtain T∗ = 9925061, I∗ = 2472, V ∗ = 10000, A∗ = 49 and the Jacobian matrix evaluated at this equilibrium is :
 The eigenvalues are −4.95 × 102, −3.27 × 10−2, −3.27 × 10−3 ± 0.022i. All real parts are negative thus equilibrium is stable.
The eigenvalues are −4.95 × 102, −3.27 × 10−2, −3.27 × 10−3 ± 0.022i. All real parts are negative thus equilibrium is stable.
A.5 Dynamical aspects
The equilibrium analysis in the previous sections does not yet tell the full story of the evolution of the system (1)-(4). Depending on the parameters, a common behavior is the following: initially A will increase as response to V being above threshold V t; the increase of A will drive both I and V to zero. Such a dynamics is stable over a long period and in practice I and V will keep small values for a time long enough to ensure virus clearance (when V is small enough, due to the random nature of the events, V will disappear).
Taking I and V to be constant equal to zero, the new evolution is:

 Note that equations for I and V are missing because if the initial states are V (0) = I(0) = 0 then V (t) = I(t) for all t ≥ 0. This evolution drives T to Λ/µ and A to zero. If however during the slow decay of A a challenge is presented in the form of a virus load V > σ/a a new infection will start and V and I will rise again.
Note that equations for I and V are missing because if the initial states are V (0) = I(0) = 0 then V (t) = I(t) for all t ≥ 0. This evolution drives T to Λ/µ and A to zero. If however during the slow decay of A a challenge is presented in the form of a virus load V > σ/a a new infection will start and V and I will rise again.
In conclusion, the stable equilibrium (20)-(21) is not necessarily reached in practice. The precise dynamics depends crucially on the parameters b and β1, see main text for details.
B Choice of simulation parameters
Parameters’ order of magnitude were derived from literature. Parameters were then fitted to SARS-CoV-2 clinical data. The value of the parameter ‘µ’ which represents the death rate of uninfected epithelial cells has been estimated for influenza host-virus dynamics which also targets lung epithelial cells, [40]. We estimated the lifespan of epithelial cells to be around one month therefore µ to be equal to 0.033 day−1. As mentioned before, the initial number of target cells is  . We assumed the initial number of epithelial cells to be around 106 which leads to 3.3 × 104 cells/day for Λ. Parameter ω was estimated by a graphical analysis of viral load temporal profiles in François-Xavier Lescure et al study, [32]. The mean peak viral load among five patients was taken to compute ω. The order of magnitude of this value was graphically estimated at 106copies/1000 cells. Therefore, the production rate of free virions was determined as 103 virions per infected cells per day. We set ω to 2.0 × 103 virions per infected cells per day. Clearance rate was restricted between 1 and 10 day−1, [37],[41], and set to 3 in our simulation. The median duration of the disease for mild cases was considered to be 10 days. Hence, the lifespan of infected cells δ was estimated to be 10−1day−1, which is consistent with equation (12). Finally, antibody clearance rate was estimated by the results of recent studies, [34], [35] which reported that IgG started to decrease in patients within 2-3 months after infection. Therefore, we assume σ to be equal to
. We assumed the initial number of epithelial cells to be around 106 which leads to 3.3 × 104 cells/day for Λ. Parameter ω was estimated by a graphical analysis of viral load temporal profiles in François-Xavier Lescure et al study, [32]. The mean peak viral load among five patients was taken to compute ω. The order of magnitude of this value was graphically estimated at 106copies/1000 cells. Therefore, the production rate of free virions was determined as 103 virions per infected cells per day. We set ω to 2.0 × 103 virions per infected cells per day. Clearance rate was restricted between 1 and 10 day−1, [37],[41], and set to 3 in our simulation. The median duration of the disease for mild cases was considered to be 10 days. Hence, the lifespan of infected cells δ was estimated to be 10−1day−1, which is consistent with equation (12). Finally, antibody clearance rate was estimated by the results of recent studies, [34], [35] which reported that IgG started to decrease in patients within 2-3 months after infection. Therefore, we assume σ to be equal to  . β0,a,b and β1 were adjusted to best fit clinical data.
. β0,a,b and β1 were adjusted to best fit clinical data.
C Sensitivity with respect to parameters
We investigate here the sensitivity of the outcomes with respect to the two main parameters, the neutralizing capacity and the ADE size and we include here for completeness, some simulations evoked in the main text.
Acknowledgements
Ghozlane Yahiaoui is supported by the Engineering and Physical Sciences Research Council (EPSRC): CDT Grant Ref. EP/L015811/1.
Footnotes
↵* The order of the authors is alphabetic
E-mail address: Antoine.Danchin{at}normalesup.org
E-mail address: Oriane.Pagani-Azizi{at}espci.fr
E-mail address: Ghozlane.Yahiaoui{at}maths.ox.ac.uk
References



