
Abstract
Worldwide, we are currently in an unprecedented situation with regard to the SARS-Cov-2 epidemic, where countries are using isolation and lock-down measures to control the spread of infection. This is a scenario generally not much anticipated by previous theory, and in particular, there has been little attention paid to the question of extinction as a means to eradicate the virus; the prevailing view appears to be that this is unfeasible without a vaccine. We use a simple well-mixed stochastic SIR model as a basis for our considerations, and calculate a new result, using branching process theory, for the distribution of times to extinction. Surprisingly, the distribution is an extreme value distribution of the Gumbel type, and we show that the key parameter determining its mean and standard deviation is the expected rate of decline ρe = γ(1 − Re) of infections, where γ is the rate of recovery from infection and Re is the usual effective reproductive number. The result also reveals a critical threshold number of infected I† = 1/(1 − Re), below which stochastic forces dominate and need be considered for accurate predictions. As this theory ignores migration between populations, we compare against a realistic spatial epidemic simulator and simple stochastic simulations of sub-divided populations with global migration, to find very comparable results to our simple predictions; in particular, we find global migration has the effect of a simple upwards rescaling of Re with the same Gumbel extinction time distribution we derive from our non-spatial model. Within the UK, using recent estimates of ≈ 37,000 infected and Re ≈ 0.9, this model predicts a mean extinction time of 616 ± 90 days or approximately ~ 2 years, but could be as short as 123 ± 15 days, or roughly 4 months for Re = 0.4. Globally, the theory predicts extinction in less than 200 days, if the reproductive number is restricted to Re < 0.5. Overall, these results highlight the extreme sensitivity of extinction times when Re approaches 1 and the necessity of reducing the effective reproductive number significantly (Re ≲ 0.5) for relatively rapid extinction of an epidemic or pandemic.
INTRODUCTION
The SIR model has remained a popular paradigm to understand the dynamics of epidemics1,2, despite its simplifications compared to real world epidemics, which have spatial structure3,4 and heterogeneity5,6 in connection between regions. But it is this simplicity that distills the course of an epidemic down to a few key parameters that gives it its power. One such parameter is the reproductive number, which is a dimensionless number that describes whether an epidemic is growing (Re > 1) or shrinking (Re < 1) in a population, and encapsulates the number of secondary infections within the period of infectivity (1/γ). Simple models can therefore be very instructive on qualitative behaviour, particularly when the parameters of such models are interpreted as effective and coarse-grained representations of more detailed models. These detailed models are necessary for quantitative accurate predictions, but such highly parameterised models can suffer from a lack of transparency in understanding the effect on the dynamics of changing different parameters6,7. For this reason, simplified coarsegrained models, such as the SIR model, have the major advantage of being highly transparent, particularly when analytical solutions are available. Both approaches used together provide a powerful approach to prediction in epidemics.
This paper examines the SIR model, but fully accounting for the discreteness of individuals, that leads to stochasticity in the progress of an epidemic. Why do we need to worry about stochastic effects in an epidemic, and in particular, to understand extinction? A key danger of deterministic models, whether we treat as continuous the number of infected individuals or the density of infected individuals, is the infinite indivisibility of a population. Particularly, in the latter case, if we consider a continuous model of isolation or lockdown and potential rebound (second wave), then there is danger of the model giving densities that actually correspond to much less than 1 individual in the population — a physical impossibility; unchecked this model will incorrectly predict regrowth of infections once restrictions are relaxed7. In a continuous model, the problem is what to do when the number of infected corresponds to less than 1 individual, and what is really required is a full stochastic description, as we do in this paper; in a stochastic model there can be exactly 0 individuals in a population, so unless there is migration of cases, the epidemic will be extinct in that population once there are 0 infected, and there can be no rebound or second wave.
There has been a considerable amount of work done to understand stochastic aspects of epidemics8 from understanding critical community sizes in diseases such as measles9,10, stochastic phases in the establishment of epidemics11, to stochastic extinction. With regard stochastic extinction most results have been focussed on understanding the time to extinction either through the whole course of an epidemic12,13 or assuming a quasiequilibrium has been reached through herd immunity in the population14. However, the situation currently faced in many countries has not been considered in previous modelling, that is the situation where significant herd-immunity has not been achieved in the population, and isolation measures have led to the reproductive number being reduced to less than 1, the critical threshold for growth.
It is with this scenario in mind, where a population still has many more susceptible (& recovered), compared to infected, where the main results of the paper are focussed, and we find the stochastic dynamics tractable within a simple birth-death branching process framework. Our main theoretical result is the distribution of the times to extinction of an epidemic, which surprisingly, we find is a Gumbel-type extreme value distribution. Key to this result is a new threshold I† = 1/(1 − Re), where f indicates extinction, below which stochastic changes dominate; when Re > 0.6 and approaches one, we show this gives significantly shorter mean extinction times compared to deterministic predictions. As this result ignores spatial structure and heterogeneity of an epidemic, we then compare to simple and more complex spatial epidemic simulations, and find our theory captures the extinction time distribution very well, as long as Re is appropriately rescaled to account for migration. We then use this theory to make broad predictions of extinction times within the UK, and globally to serve as a guide to more complex and detailed models. Our key message is that for reproductive numbers Re > 0.6 and approaching one, extinctions times increase very rapidly, and are of order many years, but can very quickly drop to times much less than a year or a few months if restricted to Re < 0.5.
SUSCEPTIBLE-INFECTED-RECOVERED (SIR) MODEL OF EPIDEMIOLOGY
The SIR model divides the population of N individuals in a region into 3 classes of individuals: susceptible S (not infected and not immune to virus), I infected and R recovered (and immune, so cannot be re-infected). If we assume a rate β of an infected individual infecting a susceptible individual (S + I → I), and a rate γ that an infected person recovers from illness (S → R), the ordinary differential equations describing the dynamics of this process are:

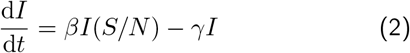

The most important thing about this model is that it allows a simple characterisation of when number of infections will grow or decline: whatever the previous history of the epidemic, for growth we need  and this happens for the following condition on RHS of the 2nd equation above: βS(t)/N − γ > 0, or equivalently,
and this happens for the following condition on RHS of the 2nd equation above: βS(t)/N − γ > 0, or equivalently,  where we have defined the dimensionless number Re as the combination shown, and will in general be time-dependent, as the number of susceptible individuals in a population change. Re represents the average number of individuals an infected person infects through the duration of the infection τ = 1/γ. It is important to understand that this interpretation of Re is within the context of a well-mixed model. In reality, locally there may be deviations from the global density of susceptible individuals (S(t)/N) and also differences in connectivity between different regions causing differing rates of infection locally. The parameter β is the rate per infectious individual of causing a successful secondary infection. It is useful to decompose this parameter as β = να, where v is the rate of making contacts per individual and α is the probability of each contact causing an infection; the probability of infection α is largely controlled by the biology of the virus/pathogen, whilst the rate of making contacts ν is mainly controlled by human behaviour, and so interventions like social distancing, lock-downs and track, trace and isolate will have an aggregate effect on ν, by reducing the rate of contacts between individuals.
where we have defined the dimensionless number Re as the combination shown, and will in general be time-dependent, as the number of susceptible individuals in a population change. Re represents the average number of individuals an infected person infects through the duration of the infection τ = 1/γ. It is important to understand that this interpretation of Re is within the context of a well-mixed model. In reality, locally there may be deviations from the global density of susceptible individuals (S(t)/N) and also differences in connectivity between different regions causing differing rates of infection locally. The parameter β is the rate per infectious individual of causing a successful secondary infection. It is useful to decompose this parameter as β = να, where v is the rate of making contacts per individual and α is the probability of each contact causing an infection; the probability of infection α is largely controlled by the biology of the virus/pathogen, whilst the rate of making contacts ν is mainly controlled by human behaviour, and so interventions like social distancing, lock-downs and track, trace and isolate will have an aggregate effect on ν, by reducing the rate of contacts between individuals.
ASSUMPTION OF CONSTANT Re WITH SMALL FRACTION OF INFECTED INDIVIDUALS IN SIR MODEL
In an idealised SIR epidemic, if 0 does not change due to behaviourial changes, the reproductive number is in general a constantly decreasing number, due to the susceptible pool of individuals diminishing – this is what would eventually lead to herd immunity as the decreasing susceptible fraction brings Re < 1. However, changes in social behaviour can also bring Re < 1 by controlling 0, before any significant herd immunity is established, which is currently the case in many countries. In this case, if we assume that the fraction of the population that have been infected is small, we can assume that the effects of herd immunity are negligible.
Estimates by the UK Office for National Statistics15 (June 12th 2020), estimate from serological testing in England that 6.8% of the population have been infected (≈ 4.6 × 106) and from random PCR testing a current incidence of 0.055% (≈ 3.7 × 104 actively infected in UK) – in which case, extrapolating to a UK population size of ≈ 67 × 106, the total number of susceptibles (≈ 62 × 106) is much greater than the number currently infected (as of 9th Aug 2020 there are more recent estimates but these numbers have not changed significantly).
In this case, as long as Re isn’t very close to 1, it is reasonable to assume that the population of susceptible individuals S(t) = S0 is roughly constant and the reproductive number unchanging  ; although each time an individual is infected, we loose exactly one susceptible the relative change of the susceptible pool is negligible, since the total number of susceptible individuals is very large. It can be shown that this approximation is good as long as
; although each time an individual is infected, we loose exactly one susceptible the relative change of the susceptible pool is negligible, since the total number of susceptible individuals is very large. It can be shown that this approximation is good as long as  , which corresponds to when the decline is sufficiently rapid, for the initial value of Re, that the error due to ignoring the change in the susceptible pool is negligible; for I0 = 3.7 × 104 and S0 = 62 × 106, we expect this approximation to be good for 1 − Re ≫ 0.024. The last differential equation involving R is really only there for book-keeping, as there is no direct effect of R on the dynamics of S and I – even with a more complicated model where immunity only lasts a finite time (SIRS), thereby increasing the susceptible pool again, if we assume the total number of susceptibles is very large in comparison, these effects will be negligible. This means for the case where only a small fraction of the population are ever infected, the SIR dynamics results in a single differential equation for I(t):
, which corresponds to when the decline is sufficiently rapid, for the initial value of Re, that the error due to ignoring the change in the susceptible pool is negligible; for I0 = 3.7 × 104 and S0 = 62 × 106, we expect this approximation to be good for 1 − Re ≫ 0.024. The last differential equation involving R is really only there for book-keeping, as there is no direct effect of R on the dynamics of S and I – even with a more complicated model where immunity only lasts a finite time (SIRS), thereby increasing the susceptible pool again, if we assume the total number of susceptibles is very large in comparison, these effects will be negligible. This means for the case where only a small fraction of the population are ever infected, the SIR dynamics results in a single differential equation for I(t):
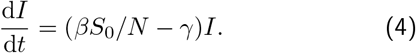
The solution to this is of course an exponential function:
 where ρe = γ(Re−1) is the effective growth rate for Re > 1, and decay rate when Re < 1, and I0 = I(0) the initial number of infected individuals. Note that Re is not a rate, it does not, in absolute terms, tell you anything about the time scales of change; however, ρe is a rate, and if it could be measured empirically, it would give information in the speed of spread of the infection, as well as having the same sign information for the direction of change (ρe > 0 the epidemic spreads, while ρe < 0 means the epidemic cannot spread).
where ρe = γ(Re−1) is the effective growth rate for Re > 1, and decay rate when Re < 1, and I0 = I(0) the initial number of infected individuals. Note that Re is not a rate, it does not, in absolute terms, tell you anything about the time scales of change; however, ρe is a rate, and if it could be measured empirically, it would give information in the speed of spread of the infection, as well as having the same sign information for the direction of change (ρe > 0 the epidemic spreads, while ρe < 0 means the epidemic cannot spread).
We are interested in understanding extinction of an epidemic and so from here on we define the rate ρe = γ(1 − Re) to be a positive quantity, making the assumption that Re < 1. In this case we can make a simple deterministic prediction for the time to extinction, by calculating the time for the infected population to reach I(t) = 1:

Of course, we want to know the time to complete elimination I(t) = 0, but we cannot answer this question with a deterministic continuous approximation, since the answer would be ∞; the time it takes to go from 1 infected individual to 0 cannot be handled in a deterministic approach, since it ignores the discreteness of individuals and the stochasticity that lies therein. In fact, without understanding the stochasticity of the extinction process, it is difficult a priori to say anything about the goodness of this deterministic calculation, since in general we would expect stochasticity to be important far before there remains only a single infected individual.
STOCHASTIC EXTINCTION OF AN EPIDEMIC
The above analysis assumes deterministic dynamics with no discreteness – it ignores any randomness in the events that lead to changes in number of infected individuals; an infected person might typically take the tube to work, potentially infecting many people, whilst on another day decide to walk or take the car, reducing the chances of infecting others. When the epidemic is in full flow with large numbers of individuals infected, all the randomness of individual actions, effectively average out to give smooth almost deterministic behaviour. However, at the beginning of the epidemic, or towards the end, there are very small numbers of individuals that are infected, so these random events can have a large relative effect in how the virus spreads and need a stochastic treatment to analyse. We are interested in analysing the stochasticity of how the number of infected decreases when Re < 1 and eventually gives rise to extinction, i.e. when there is exactly I = 0 individuals; in particular, we are primarily interested in calculating the distribution of the times to extinction.
We can initially confirm that the assumptions of a constant Re due to a negligibly changing susceptible population of the previous section are accurate, by running multiple replicate stochastic continuous time simulations with Poisson distributed events (known as Gillespie or kinetic Monte Carlo simulations)16 of the SIR model with Re = 0.7, γ = 1/7 days−1, I0 = 3.7 × 104 and an initial recovered population of R(0) = 6 × 106 (for simplicity we take 10% infected and recovered as a more conservative estimate than from the serological surveillance data in the UK15). Fig.1 plots the decline in number of infected over time I(t). Each of the trajectories from the Gillespie simulations is a grey curve, whilst the deterministic prediction (Eqn.5) is shown as the solid black line. We see that for I(t) ≫ 1 the stochastic trajectories are bisected by the deterministic prediction, indicating that the assumption of a constant Re is a good one.

Simulation trajectories on log-linear scale (inset: linear-linear scale) for a decay rate of ρe = 0.043/day, corresponding to Re = 0.7, 1/γ = 7 days, I0 = 3.7 × 104 and an initial recovered population of R(0) = 6 × 106. The solid black line is the deterministic prediction from Eqn.5, grey trajectories are 100 replicate Gillespie simulations of a standard SIR model, whilst the yellow trajectories are from 50 replicates using the spatial epidemic simulator GleamViz17,18 restricted to the United Kingdom with a gravity model between heterogeneous sub-populations as shown in the inset map of the UK.
Heuristic treatment
We can see from Fig.1 that as I(t) approaches extinction, as expected the trajectories become more and more varied as the number of infected becomes small. A simple heuristic treatment inspired from population genetics19 would define a stochastic threshold I†, below which stochastic forces are more important than deterministic; the time to extinction is then a sum of the time it takes to go deterministically from I0 to  and the time it takes to go from I† to I = 0 by random chance.
and the time it takes to go from I† to I = 0 by random chance.
Assuming such a threshold I† exists, this latter stochastic time can be approximated as follows: if there are I† individuals and changes are mainly random, then we are randomly drawing individuals from a pool of I† infected individuals and N − I† non-infected individuals — a binomial random walk — which when I† ≪ N has standard deviation ≈ I† per random draw, which means we need k = I† random draws, such that the standard deviation over those k draws is  ; a single random draw corresponds to one infection cycle of the virus, which is τ = 1/γ days, so the time to extinction starting with I† individuals is approximately I†/γ.
; a single random draw corresponds to one infection cycle of the virus, which is τ = 1/γ days, so the time to extinction starting with I† individuals is approximately I†/γ.
How do we estimate I†? It is given by the size at which random stochastic changes, change the number of infected by the same amount as the deterministic decline. In one cycle or generation of infection, if there was no stochasticity, the number of infected would decline by ≈ ρeI†/γ, so equating this to the expected standard deviation of purely random changes,  , we find I† = 1/(1 − Re). As discussed below, and in more detail in the Appendix, a more exact calculation of these considerations, using branching processes, gives exactly the same expression for I†. This means the typical stochastic phase lasts
, we find I† = 1/(1 − Re). As discussed below, and in more detail in the Appendix, a more exact calculation of these considerations, using branching processes, gives exactly the same expression for I†. This means the typical stochastic phase lasts  and so adding the deterministic and stochastic phases, the mean time to extinction
and so adding the deterministic and stochastic phases, the mean time to extinction  (see Eqn.8 below for a more exact expression of the mean).
(see Eqn.8 below for a more exact expression of the mean).
Exact branching process analysis
The branching process framework used to calculate the distribution of extinction times is standard, but detailed, and so we will sketch the derivation here and leave details for Appendix 1. The first step is to recognise that there are two independent stochastic events that give rise the net change in the numbers of infected individuals, as depicted in Eqn. 4 for the continuum deterministic limit: 1) a susceptible individual is infected by an interaction with a infected individual, such that I → I +1 and 2) an infected individual recovers spontaneously such that I → I − 1. This is a simple birth death branching process for which it possible to write down differential equations (dpI (t)/dt) for how the probability of I infected individuals changes with time in terms of the birth and death events just defined. It is possible to find after some calculation the probability generating function G(z, t) of the birth-death process, from which the probability of having exactly I = 0 individuals as a function of time is given by:
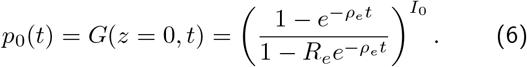
If p†(t) is the distribution of times to extinction (i.e. the probability of an extinction occuring between time t and t +dt is p†(t)dt), then clearly the integral of this distribution, between time 0 and t is exactly Eqn.6, and hence the distribution of times to extinction is simply the derivative of p0(t) with respect to time. Doing this and also taking the limit that I0 ≫ I†, we find:
 where
where  , which is the time it takes for number infected to change from the initial number I0 to the critical infection size, which this calculation shows is given by
, which is the time it takes for number infected to change from the initial number I0 to the critical infection size, which this calculation shows is given by  , which is the same result as arrived by the heuristic analysis above. Fig.2 shows a histogram (grey bars) from Gillespie simulations of the SIR model with 5000 replicates of the number of infected individuals for Re = 0.7, γ = 1/7 days, I0 = 3.7 × 104 and an initial recovered population of R(0) = 6 × 106 and the corresponding prediction from Eqn.7 (solid black line) — we see that there is an excellent correspondence.
, which is the same result as arrived by the heuristic analysis above. Fig.2 shows a histogram (grey bars) from Gillespie simulations of the SIR model with 5000 replicates of the number of infected individuals for Re = 0.7, γ = 1/7 days, I0 = 3.7 × 104 and an initial recovered population of R(0) = 6 × 106 and the corresponding prediction from Eqn.7 (solid black line) — we see that there is an excellent correspondence.

Probability density of extinction times for the same parameters as in Fig.1. Grey bars are a histogram of 5000 replicate simulations of Gillespie simulations normalised to give an estimate of the probability density, and the black curve is the prediction of the analytical calculation given in Eqn.7, which we see matches the simulations extremely well. The yellow bars are histograms from the GleamViz spatial epidemic simulator with 50 replicates, which we see gives similar results to the predictions of the stochastic SIR model.
Surprisingly, the extinction time distribution is a Gumbeltype extreme value distribution20,21; it is surprising as an extreme value distribution normally arises from the distribution of the maximum (or minimum) of some quantity, although here it is not clear how this relates to the extinction time. There are number of standard results for the Gumbel distribution Eqn.7, so we can write down (or directly calculate) the mean and standard deviation of the extinction time:

 where Υ ≈ 0.577 is the Euler-Mascheroni constant. We see that the heuristic calculation overpredicts the stochastic part of the extinction time by a factor of ≈ 2. Note that the standard deviation or dispersion of the distribution only depends on the inverse of the rate of decline ρe and as expected not on the initial number of infected individuals I0; hence as ρe decreases (Re gets closer to 1), we see that the distribution of extinction times broadens (as we see below in Figs.4&6).
where Υ ≈ 0.577 is the Euler-Mascheroni constant. We see that the heuristic calculation overpredicts the stochastic part of the extinction time by a factor of ≈ 2. Note that the standard deviation or dispersion of the distribution only depends on the inverse of the rate of decline ρe and as expected not on the initial number of infected individuals I0; hence as ρe decreases (Re gets closer to 1), we see that the distribution of extinction times broadens (as we see below in Figs.4&6).
We can also calculate the cumulative distribution function
 from which the inverse cumulative distribution function T† = (P†)−1 can be calculated:
from which the inverse cumulative distribution function T† = (P†)−1 can be calculated:
 which enables direct generation of random numbers drawn from the extinction time distribution, by drawing uniform random u on the unit interval and calculating T†(u). It also allows calculation of arbitrary confidence intervals, for example, the 95% confidence intervals, by calculating T†(0.025) and T†(0.975), as well as the median
which enables direct generation of random numbers drawn from the extinction time distribution, by drawing uniform random u on the unit interval and calculating T†(u). It also allows calculation of arbitrary confidence intervals, for example, the 95% confidence intervals, by calculating T†(0.025) and T†(0.975), as well as the median 
 .
.
Finally, it is important to stress that the distribution of extinction times Eqn.7 and the following results all assume that I0 » I†, so that there is a clear separation of the deterministic and stochastic phases of the decline in infections. A more general and exact result for the distribution of extinction times is given in Appendix 1.
EXTINCTION TIME DISTRIBUTION WITH SPATIAL STRUCTURE AND HETEROGENEITY
National level (United Kingdom)
A potentially valid criticism is that real populations have spatial structure and heterogeneity of contacts between regions. To make comparison to our simple predictions, we used a complex epidemic simulator GleamViz (v7.0)17,18, which includes a gravity model of migration, where rates of migrations between sub-populations are proportional to their population sizes (see Fig.1 inset map of UK), and each sub-population based on accurate census data within a grid of 25 km. We ran 50 replicate simulations for an SIR epidemic within the United Kingdom and with zero air travel to other countries, with the same parameters as the stochastic SIR simulations in the previous section (Re = 0.7, γ = 1/7 days, initial recovered population R(0) = 6 × 106 – in addition, each definable sub-population in the UK was given a current infection incidence of 0.06% giving a total I0 ≈ 3.7 × 104). We see the trajectories (Fig.1 – yellow lines) and histogram of extinction times (Fig.2 – yellow bars) compare very favourably to the predictions of the stochastic SIR model (black solid line and grey histogram bars); the mean and standard deviation including the gravity migration model is 211 ± 16 days, which is slightly smaller than the prediction of the stochastic SIR model which has no migration or spatial structure (231 ± 30 days). This suggests that heterogeneity and migration might together have the net effect of reducing extinction times, as below we see increasing migration uniformly, has the opposite effect; nonetheless within the UK it would seem the overall effect of heterogeneity and migration is of second order to predictions of a well mixed model. Overall, at a national level, we find the results of our simple model are accurate to within the width of the distributions of the extinction times.
Global
It was not possible to repeat these simulations on a global scale as GleamViz does not record individual level changes in infections and deaths in its global output. Here instead we first consider the total extinction time distribution for a number of isolated regions (countries) with no migration between, but each with the same Re. As we show in Appendix 2, in fact, the extinction time distribution of the whole region (i.e. the distribution of the maximum time of all the groups) is exactly the same distribution as assuming a single unstructured/undivided population for the region. We verify this by Gillespie simulation of a simple birth-death model with growth rate γRe and death rate γ for n isolated populations; the grey histogram in Fig.3 is the estimate of the extinction time distribution for isolated sub-populations and this matches the grey solid line, which is exactly the solid black line in Fig.2.
We now look at the effect of migration, where we examine the same Gillespie simulations of birth and death, but with a probability of global migration per individual of ϕ. As we increase ϕ we see that the extinction time distribution shifts to longer times, yet still maintains the same form as given by Eqn.7 – fitting to this equation using only Re as a free parameter, we find for ϕ = {0.01, 0.05, 0.1}, Ȓe = {0.709±0.001, 0.732±0.001, 0.760 ±0.001}, respectively (minimum R-sqd statistic of 0.975). These fits are shown as the blue and red solid lines in Fig.3 for ϕ = 0.05 and ϕ = 0.1, respectively, and we see that the fits follow the data very closely (the histogram and fits for ϕ = 0.01 are not shown in Fig.3 for clarity, as they overlap closely with ϕ = 0). We can see that can predict these estimated reproductive numbers Re by simply rescaling the base Re to Re → (1 + ϕ)Re = {0.707, 0.735, 0.77} for ϕ = {0.01, 0.05, 0.1}, respectively. This finding is closely related to the literature on the group level reproductive number R*3–5, except here we are studying the decline and extinction of an epidemic/pandemic as opposed to its establishment, which has not been previously studied in this context. Overall, these results suggest that under the assumption that each national region has the same Re, that the extinction time distribution is given by the stochastic SIR model (Eqn.7) but with a rescaled Re to account for air traffic or migration between regions/countries.

Probability density of extinction times for the same parameters as in Fig.1, but including migration and sub-division into equal sized populations. Each histogram comprises 1000 replicates for n = 5 regions connected by uniform migration with probability ϕ. Grey bars are ϕ = 0 (complete isolation), blue correspond to ϕ = 0.05 and ϕ = 0.1 are the red bars. For ϕ = 0 the solid line grey line is exactly the solid black line in Fig.2, showing that the extinction time distribution of identical to the single global well-mixed population of same aggregate size. The solid blue and red lines are fits to the histogram using Eqn.7 with a single free parameter Re (with γ and I0 constrained to the values used to run the simulations.
EXTINCTION TIME PREDICTIONS
United Kingdom
We first consider what this model predicts for the extinction of the SARS-Cov-2 epidemic within the United Kingdom, given an incidence of 0.055% (12th June 2020 — a more recent estimate show this incidence has not changed significantly) or I0 ≈ 37 × 104 were infected at the beginning of June15. In Fig.4 we have plotted the estimates of mean (solid lines), median (dashed lines) and deterministic (Eqn.5 – dots) extinction times, together with 95% confidence intervals (shaded region), given an initial number of infected I0 for various reproductive numbers Re between 0 <Re < 1, and for various values of γ.

Prediction of the extinction times from analytical theory of the epidemic within the United Kingdom as a function of Re with an initial infected population of I0 = 3.7 × 104 for various values of the recovery rate: 1/γ = 7 days (yellow); 1/γ = 10 days (red); 1/γ = 20 days (green). Solid lines are the predictions of the mean (Eqn.8), dashed lines the median, the dots correspond to the deterministic prediction (Eqn.5), whilst each shaded region corresponds to the 95% confidence interval; the median and confidence intervals are calculated directly from Eqn.11.
We see three broad trends: 1) for R>≈ 0.6 the extinction times are very long of order years, whilst at the same time the 95% confidence intervals becomes increasingly broad (of order years themselves); 2) also for Re >≈ 0.6, the deterministic prediction increasingly and significantly overestimates the mean extinction time; 3) for Re < 0.5 the extinction times plateau with diminishing returns for further decreases in the reproductive number; and 4) as the mean infection duration 1/γ increases the extinction times increase and the 95% confidence intervals go up in proportion. Regarding point 3), we see that there is minimum time to extinction, given by Re = 0; from Eqn.8 in the limit that Re → 0, the mean time to extinction 〈t〉 → 1/γ(ln(I0)+Υ), which shows the extinction process is ultimately limited by the rate of recovery γ, imposing a maximum speed limit on the rate of decline of infections.
To make specific predictions, we need to estimate the recovery rate γ. Current direct estimates for the mean or median infection durations are quite broad, and also in need of careful interpretation; different individuals will have very different disease progressions and also very different transmission potentials, from those that are asymptomatic to individuals that are hospitalised. A recent comprehensive survey of the literature22 looked at three different types of infection durations: 1) asymptomatic (T2); 2) pre-symptomatic (T3) and 3) symptomatic (T5). Their survey suggest a median T2 of ≈ 7 days (4−9.5), a mean T3 of 2 days, and a median T5 of 13.4 days. On the other hand an early study of time to recovery of patients within and outside of China, suggests 1/γ ~ 20 days23. Overall, it is arguable that the T2 time is likely to be most relevant, since infection events are likely to be dominated by asymptomatic carriers, and symptomatic times T5 in hospital are likely to be an overestimate of the duration for which an individual has high transmission potential. In addition, as suggested by this study the T5 time is typically estimated by time to a negative RT-PCR test, but the time when the individual had viral loads sufficient to infect is likely to much shorter. However, the scope of this paper is not to make very precise predictions, but to demonstrate broad trends, and so as well as plotting predictions for what seems to be the most likely infection duration 1/γ = 7 days (yellow), we have plotted predictions for 1/γ = 10 days (red) and 1/γ = 20 days (green). In general, we see that longer infection durations give rise to longer extinction times, since as mentioned above the recovery rate sets the overall tempo for the decline in infections.
Assuming a duration of 1/γ = 7 days, we see that the simple stochastic SIR model predicts that extinction or elimination of the epidemic can occur within the United Kingdom of order about 100 days or a few months, if Re can be kept to below about 0.5. More precisely, if Re = 0.4, the mean time is 123 ± 15 days with 95% confidence intervals:(102, 160) days. However, as of the end of June, both the UK government’s own estimate (SPI-M modelling group)24 and the MRC Biostatistics Unit, Cambridge25 estimated a reproductive number put Re ≈ 0.9 for the region of England, which gives a mean time of 616 ± 90 days (484, 832), or roughly two years; as Re = 0.9 is close to a value of the reproductive number where would expect our underlying assumptions of an unchanging susceptible population to be less good (
 ), we checked this prediction by Gillespie simulation of the SIR model, and find a mean extinction time of 592 ± 85 days, which is slightly less than our prediction, but with a difference which is still within the spread of times we would expect in reality.
), we checked this prediction by Gillespie simulation of the SIR model, and find a mean extinction time of 592 ± 85 days, which is slightly less than our prediction, but with a difference which is still within the spread of times we would expect in reality.
Estimating extinction times from direct estimate of ρe
Precise predictions of extinction times based on Eqn.7, as shown in Fig.4, require knowledge of both Re and γ, which are generally quite difficult to estimate. However, Eqn.7 is mainly dependent on the rate of decline of the epidemic ρe, with weak dependence separately on Re and γ. ρe can be determined more straightforwardly if we assume current daily deaths are proportional to the number infected; if infections are declining exponentially at a rate ρe then so will the number of daily deaths, so a curve fit will give an accurate measure of ρe even if we cannot determine the proportionality constant to translate deaths to infections. An alternative, could be to look at time-series of number of daily infections per number of tests performed, to remove biases due to testing, however, here the aim is to illustrate why a direct estimate of ρe is useful, rather than to estimate this number very accurately.
As shown in Fig.5, fitting decaying exponentials to daily number of deaths (date of death, not date of reporting) from the NHS UK26, from the moment of decline to 13th July 2020, shows a very good fit (showing deaths are declining as a simple exponential and giving further weight to our simple model), giving an England wide estimate of ρe = 0.043 ± 0.001 days−1, with a range of ρe = 0.036±0.001 days−1 (slowest decline) for North York-shire and ρe = 0.069±0.001 days−1 (most rapid decline) in London. Using the rate of decline of England, we estimate a mean extinction time in the UK as 231±30 days (95% CI: (187, 303) days), for Re = 0.7, 1/γ = 7 days and 238 ± 30 days (95% CI:(195, 310) days), for Re = 0.57, 1/γ = 10 days; as we can see changing Re and γ for a fixed ρe does not change the predictions significantly, and we suggest in general, determining ρe could be a more robust way to estimate extinction times.

Data from NHS England26 on daily deaths within hospitals in England and different regions within England. Each region is fit using a simple decaying exponential from 40 days after the 1st March 2020, where each region first shows a clear decline in number of daily deaths.
These estimates are much shorter than the approximately 2 years estimated above using separate estimates of Re and γ to calculate ρe. It is clear that in some of the regions, there is relatively poor fit at later times, with an indication that overall in England the rate of decline ρe is decreasing, which could account for the difference in estimates. However, a more careful probabilistic analysis6 is required to support such a conclusion, which is outside the scope of this paper; as the above stochastic calculation and simulations show, as the number of infections — or deaths in this case — is small, trajectories become more stochastic and varied, and importantly, can’t be simply treated as a deterministic trend plus (Gaussian) noise, which is reasonable when numbers are large. Trying to fit a more complicated model could result in fitting noise; in other words we might be wrongly concluding a decrease in ρe due to a trajectory in a region which just happens to randomly decline slightly less quickly. It is likely that the UK government’s advisory SPI-M modelling group24 does exactly this in its direct estimates of ρe (as far as the author is aware details are not publicly available), which do indicate an overall slower rate of decline with a range quoted of ρe = 0.01 → 0.04; assuming 1/γ = 7 days, we would arrive at the same estimate above for the extinction time, for Re with ρe ≈ 0.014, which is consistent with the range quoted.
Global extinction time predictions
We can also use our calculation to make approximate predictions of global extinction times of SARS-Cov-2, with all the same broad caveats, given the simplifications of the model. However, as argued above at a global level, we can have confidence that the predictions of the extinction time distribution Eqn.7 can be quantitatively correct, when the Re value is effectively rescaled to account for migration/air-traffic between nations. The current global death rate is approximately 5000 deaths per day (9th August), and so with an approximate infection fatality rate η ≈ 0.01,23,27 the rate of change of deaths would be roughly d(deaths)/dt = ηγI(t); inverting this gives a crude estimate of the current number of actively infected globally as I0 ≈ 3.5 × 106, or roughly 3.5 million. More precise estimates would require understanding the age structure of the infection fatality rate, which is highly biased to older populations. Given a global population of 7.8 billion this corresponds to a global incidence of ≈ 0.05%, which is very similar to that measured more directly within the UK. In Fig.6 we plot how the predicted extinction time changes with effective reproductive numbers between 0 <Re < 1 for 1/γ = 7 days (yellow), given I0 = 3.5 × 106, and also for 1/γ = 10 days (red) and 1/γ = 20 days, where we correct our initial estimate of I0 for different values of γ, giving I0 = 5 × 106, and I0 = 107, respectively. As would be expected, the results mirror the predictions for within the United Kingdom; for sufficiently small values of Re (Re < 0.5), the theory predicts global extinction on a timescale of approximately 200 days or 6 to 7 months, whilst Re > 0.6 lead to very long extinction times (~years). More precisely, for Re = 0.4 and assuming 1/γ = 7 days, we find a mean extinction time of 177 ± 15 days (95% CI: (155, 213) days).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Prediction of the extinction times from analytical theory of the pandemic globally as a function of Re, for various values of the recovery rate and initial infected corresponding to 5000 deaths globally per day (as described in main text): 1/γ = 7 days & I0 = 3.6 × 106 (yellow); 1/γ = 10 days & I0 = 5 × 106 (red); 1/γ = 20 days & I0 = 107 (green). Solid lines are the predictions of the mean (Eqn.8), dashed lines the median, the dots correspond to the deterministic prediction (Eqn.5), whilst each shaded region corresponds to the 95% confidence interval; the median and confidence intervals are calculated directly from Eqn.11.
DISCUSSION & CONCLUSIONS
We have presented a new analysis of extinction in the stochastic SIR model in the context of populations with very little herd immunity and yet a significant number of infected individuals. Using heuristic analysis we calculate the mean time to extinction and show that there is a critical threshold I† = 1/(1 − Re), below which random stochastic changes are more important, which suggests that for Re < 1, but approaching 1, a simple deterministic prediction will be poor. With a more exact branching process analysis, we then calculate in closed form the extinction time distribution of an epidemic, which, surprisingly, is an extreme value distribution of the Gumbel type, and is mainly dependent on the rate of decline of the epidemic ρe = γ(1 − Re), with a weak logarithmic dependence on Re explicitly. A key advantage of a closed form solution for the distribution is that we can discern broad trends very quickly without doing a large number of complex simulations over a large parameter space. Given the simplicity of SIR well-mixed model, we compared our results to a complex spatial epidemic simulator, GleamViz17,18 with explicit heterogeneity in connections between sub-populations, as well as simulations of uniform migration between sub-populations and find good overall agreement for the distribution of extinction times with our simple predictions.
We use these results to produce predictions of extinction times within the UK and globally of the SARS-Cov-2 epidemic/pandemic, as a function of Re as a guide to the expected trends in more complex models; the results suggest that if the reproductive number can be constrained to Re = 0.4 (assuming 1/γ = 7 days), within the UK we would expect extinction times of approximately 123±15 days, or 4 months, given an incidence of infection ≈ 0.06% and globally, roughly 177 ± 15 days, assuming a similar level of incidence. However, given current estimates of Re ≈ 0.9 within the UK, the extinction times become very long, of order a couple of years; this clearly demonstrates the sensitivity of extinction times for Re > 0.6, as shown in Figs.4&6. On the other hand the same figures show that decreasing Re much below Re = 0.4 produces diminishing gains in reductions of extinction times; hence, given social consequences of lock-down measures this suggests an optimal Re ≈ 0.4 → 0.5, which in the United Kingdom was achieved just after lock-down in regions such as London25.
However, these predictions need to be treated with care when 1 − Re becomes small, as relative changes in the susceptible pool have a large relative effect on Re, so we can no longer assume it is constant; in particular, we estimate this occurs for  , and will in general lead our extinction time estimates being an over-prediction. This does not affect our broad conclusions that for all practical purposes when Re > 0.6 the extinction times become very long and of order years. However, it is an interesting open question to calculate the extinction time when Re = 1, in the same limit that I0 ≪ S0, since infections reducing the susceptible pool now have a significant effect to reduce Re; intuitively, we might expect that Re will continue to decrease until again changes in S become negligible, such that Re stabilises and then the rest of the analysis in this paper would be valid. Note that a naive analogy to evolutionary theory where Re = 1 would correspond to neutrality, and so the epidemic could potentially increase, would be incorrect for the reasons outlined — even for Re = 1 we would expect the epidemic to decline.
, and will in general lead our extinction time estimates being an over-prediction. This does not affect our broad conclusions that for all practical purposes when Re > 0.6 the extinction times become very long and of order years. However, it is an interesting open question to calculate the extinction time when Re = 1, in the same limit that I0 ≪ S0, since infections reducing the susceptible pool now have a significant effect to reduce Re; intuitively, we might expect that Re will continue to decrease until again changes in S become negligible, such that Re stabilises and then the rest of the analysis in this paper would be valid. Note that a naive analogy to evolutionary theory where Re = 1 would correspond to neutrality, and so the epidemic could potentially increase, would be incorrect for the reasons outlined — even for Re = 1 we would expect the epidemic to decline.
Note that within the UK, throughout we have taken an incidence level of 0.055% measured by random population sampling in England between 25 May to 7 June 2020, corresponding to I0 ≈ 3.7 × 104. A more recent estimate (20 to 26 July 2020) by the ONS shows a small increase in the incidence to ≈ 0.07%, which means our estimates are still relevant as of end of July. In fact, the month of June saw a decrease to an incidence of 0.03% by random population sampling, indicating that infections levels are currently on an upward trend, which of course would be inconsistent with current government central estimates that Re < 1.
We have also used England and United Kingdom, somewhat interchangeably, purely as a matter of convenience, as the main scope is to provide approximate and qualitative predictions. The Scottish government28 estimates the current (7th Aug 2020) number of infected as I0 ≈ 200 and a weekly reduction in the infectious pool of 24%, which translates to ρe = 0.038 (Re ≈ 0.73, assuming 1/γ = 7days) giving a mean extinction time of 119 ± 33 days (95% CI: (70, 199) days); this reduces to 63 ± 15 days if ρe = 0.086, corresponding to a reproductive number of Re = 0.4. In Wales there is no direct report of currently infected, but a very crude estimate of I0 ≈ 1400 can be obtained given approximately an average of 2 deaths per day (2nd Aug → 9th Aug), an infection fatality rate of η ≈ 0.01,23,27 and 1/γ = 7 days. The rate of decline of infections is estimated to have a wide range ρe = 0.01 → 0.0829, so taking a central estimate of ρe = 0.045 (Re = 0.685, 1/γ = 7 days), we arrive at mean extinction time prediction of 148 ± 29 days (95% CI: (106, 217) days); if the rate of decline is increased to ρe = 0.086 (Re = 0.4) then this reduces to 85 ± 15 days (95% CI: (63, 121) days). In Northern Ireland, it is currently estimated Re = 0.8 → 1.830, and so is likely to be above 1, which means an extinction time estimate cannot be made; however, we can make an estimate assuming Re as for the other nations. As for Wales there is no estimate of the size of the currently infected pool, but the same crude approximation gives I0 ≈ 47, given 2 deaths in period 10th July to 9th Aug (30 days), and an extinction time estimate of 46 ± 15 days (95% CI: (24, 82) days). Again, these predictions should be viewed as a guide to expected qualitative changes in extinction times given changes in Re, and in particular, to demonstrate the sensitivity of extinction times on Re in specific local contexts. Reducing the reproductive number to Re = 0.4 is ambitious, but was achieved in London not long after the lockdown on 23rd March 202025; in other regions of England, the lockdown was less successful with numbers in the range Re = 0.6 → 0.7, which if repeated again, from Fig.4 would give an extinction time of roughly half a year for the United Kingdom as a whole.
The basic calculation in this paper ignores spatial structure3,4, heterogeneity of contacts between individuals5, and also dependence of transmission on age structure of the population. To assess the realism of our simple model, we performed simulations using a realistic spatial epidemic simulator, GleamViz,17,18 which gave a distribution of extinction times matching reasonably closely the Gumbel distribution in Eqn.7, with a slightly smaller mean time; this suggests that despite the heterogeneity of contacts between different regions, the overall decay of infection is exponential and the stochastic variation follows closely the well-mixed predictions of the stochastic SIR model presented here.
We have also made a very crude extrapolation of our results to extinction at the global level. The most problematic assumption, of course, is a single value of Re globally, and so the predictions in Fig.6 should be viewed as a guide to what could be achieved globally if all countries acted roughly in the same way on average. As we show, sub-divided regions with uniform migration, simply lead to a upwards rescaling of Re by factor (1 + ϕ) where ϕ is the migration probability, and so we can have confidence in these predictions, as long as Re at a global level is suitably interpreted. Previous work on generalising the concept of the reproductive number to include spread between different regions, uses a different approach, by defining an analogous reproductive number R* for regions,3–5, i.e. how many sub-populations or regions have at least one infection, where migration plays an analogous role to individual contacts for the spread of infection in a single population. This gives rise to a condition for spread or decline of infections to multiple regions and eventually globally, if R* > 1 or R* < 1, respectively. In the context of a local reproductive number Re < 1, as has been discussed previously3, it is still theoretically possible that R* > 1, particularly when infection is highly prevalent in a region (due to a previous time when R0 > 1, as has occurred across in many countries with SARS-Cov2 before lockdowns were imposed); this can happen if the global contact probability is sufficiently large, meaning that the infection can continue to spread between countries and regions. However, these long distance seedings of infection will not in themselves lead to regional or national outbreaks, as long as locally Re < 17; the simple upward rescaling of Re → (1 + ϕ)Re, which we observe for small ϕ captures this phenomenon. These results also suggest that at the national level, a simple rescaling of Re should describe the reduction in the rate of decline of infections due to importation of infected cases, and should be accounted in more accurate estimates of extinction times.
Finally an aspect, which we have not considered, is the possibility of a non-human reservoir of SARS-Cov-2, which could allow re-infections of human populations, such that actually extinction is not a permanent (stable) state, as we have assumed in this paper. This could be accounted in a similar way as we account for migration in this theory, except, unlike human populations (and arguably even in human populations), we have much less control, or even understanding, of a potential non-human reservoir. However, if such reservoirs, whether bat or pangolin31,32, can be identified and surveyed33, measures can be taken to control contact with human populations, such that effective global elimination in human populations is possible, even if the virus cannot be eliminated from non-human populations.
To conclude, the theory presented and closed-form analytical equation for the distribution of extinction times of an epidemic, despite the many simplifications of the SIR model, provides a useful and quick guide to estimate, with confidence intervals, the time to extinction of an epidemic; different intervention, and test and trace, strategies are encapsulated in an overall effective reproductive number Re to give extinction time predictions as shown in Figs.4&6. In particular, we have shown that these results have broad applicability, beyond the specific assumptions of the calculation, when the reproductive number is suitably rescaled to account for migration. The broad conclusion when applied to SARS-Cov-2 is that to achieve rapid extinction, on times of order or less than half a year, then the goal should be to restrict Re to numbers much less than 1 and optimally in the region Re ≈ 0.4 → 0.5.
CODE AVAILABILITY
Code to plot extinction time predictions and distributions, as well as for performing the Gillespie simulations can be found at https://github.com/BhavKhatri/StochasticExtinction-Epidemic.
ACKNOWLEDGEMENTS
I thank John McCauley (The Francis Crick Institute), Austin Burt, Vassiliki Koufopanou & Tin-Yu Hui (Imperial College) for their insights and useful comments on the manuscript.
APPENDIX 1: BRANCHING PROCESS ANALYSIS
A birth-death process with birth rate b = γRe and death rate d = γ, corresponds to a pure exponential growth (Re > 1) or decay (Re < 1) phase of an epidemic, when the number of susceptible individuals S(t) are far in excess of the total number infected I(t). In this appendix, for presentational clarity, we will use n = I to represent the number of infected individuals. To write down the rate of change of the probability of n infected individuals at time t, we need only consider the probability of having n − 1, n, n +1 individuals and rates of transitions between them, since in the limit of infinitesimal (continous) changes in time, we consider only changes of single individuals. The rate of transition from n − 1 → n happens with rate γRe(n − 1) and the rate of transition from n +1 → n happens with rate γ(n + 1), which both lead to an increase in the probability of n, while the rate of transition from n → n +1 happens with rate γRen and the rate of transition from n → n − 1 happens with rate γn, which both decrease the probability of n. Using these facts we can write down the rate of change of the probability of n at time t:
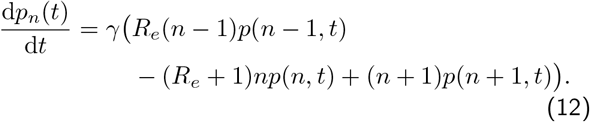
However, this description isn’t complete, and we need to consider how the probability of the n = 0 state changes, since the above equation won’t work for n − 1, since we cannot have a negative number of individuals:
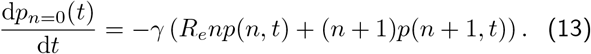
We can encompass both equations together in one by using the unit step function Un = 1 for n ≥ 0, while Un = 0 for n< 0:
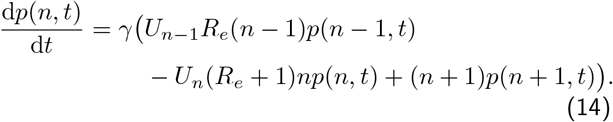
For n< 0, as long as we have an initial condition, pn<0(t = 0) = 0, the ODEs above guarantee that pn<0(t)∀t. Considering each value of n:0 ≤ n< ∞, we have an infinite set of coupled differential equations. The standard way to solve this is to use probability generating functions:
 which is in general a complex function of a complex variable z. Using the fact that z∂G(z, t)/∂t = ∑npn(t)zn, it is straightforward to show that the set of ODEs give the following first order partial differential equation for G(z, t):
which is in general a complex function of a complex variable z. Using the fact that z∂G(z, t)/∂t = ∑npn(t)zn, it is straightforward to show that the set of ODEs give the following first order partial differential equation for G(z, t):
 where
where

This PDE can be solved by using the method of characteristics, which finds a parametric path z(s), t(s) along which our original PDE is obeyed. The rate of change of G(s) along this path in terms of our parameterisation is:
 and so with reference to the original PDE (Eqn.16), we can identify that
and so with reference to the original PDE (Eqn.16), we can identify that

Integrating these pair of equations gives the characteristic paths for which dG(s)/ds = 0 is a constant:
 where C is a constant that represents different possible initial conditions. Integrating dG(s)/ds = 0, gives
where C is a constant that represents different possible initial conditions. Integrating dG(s)/ds = 0, gives
 where ϕ is an arbitrary function to be determined by consideration of the initial conditions on pn(t). We can use the fact that at time t = 0 we assume we know the exact number of infected individuals is n0 and hence,
where ϕ is an arbitrary function to be determined by consideration of the initial conditions on pn(t). We can use the fact that at time t = 0 we assume we know the exact number of infected individuals is n0 and hence,  , where
, where  for n = n0 and
for n = n0 and  for n = n0. Calculating the probability generating function for the initial delta function probability mass, we get G(z, t = 0) = zn0, and so we need to find a function ϕ satisfying:
for n = n0. Calculating the probability generating function for the initial delta function probability mass, we get G(z, t = 0) = zn0, and so we need to find a function ϕ satisfying:

Substituting x = (z − 1)/(z − 1/Re), we can find ϕ(x), to give our solution:
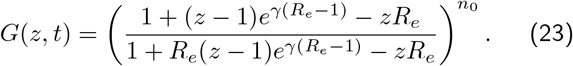
Our probability mass function pn(t), should always be normalised ∑npn(t) = G(z = 1, t) = 1; substituting z = 1 we see this that the solution G(z, t) behaves correctly. Finally, the reason this is all useful, is that we want to calculate the probability of zero individuals infected p0(t), which is simply given by G(z = 0, t), since 00 = 1:
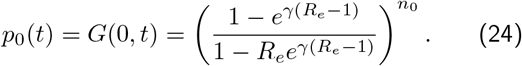
Substituting n0 = I0 and ρe = γ(1 − Re) gives Eqn.6 in the main text.
Differentiating Eqn.24 to obtain the extinction time distribution, we find
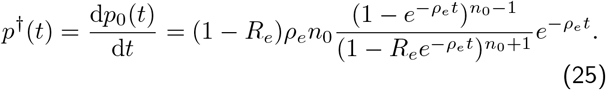
This is an exact expression, which is valid for all values of I0 and I†, as long as the original assumptions of the model that changes in susceptible numbers are negligible  is true. If I0 ≫ I† then we expect there to be a strong division between the deterministic phase and the stochastic phase, such that in Eqn.25 the exponentials have sufficiently decayed such that n0e−ρet ≪ 1, before any extinction is likely, then it is straightforward to show that the limiting form of Eqn.25, is the Gumbel distribution, Eqn.7, using the fact that
is true. If I0 ≫ I† then we expect there to be a strong division between the deterministic phase and the stochastic phase, such that in Eqn.25 the exponentials have sufficiently decayed such that n0e−ρet ≪ 1, before any extinction is likely, then it is straightforward to show that the limiting form of Eqn.25, is the Gumbel distribution, Eqn.7, using the fact that  .
.
APPENDIX 2: INVARIANCE OF EXTINCTION TIME DISTRIBUTION TO POPULATION SUB-DIVISION
If we imagine a single population to be divided into n equally sized sub-populations, each with a reproductive number Re and zero-migration between, then the extinction time distribution of tk in the kth sub-population will be given by Eqn.7, but with I0 → I0/n. Now we want to calculate the extinction time distribution of the whole population. Extinction will occur when all sub-populations have zero infected individuals. We can record the extinction times in each sub-population: t1,t2,…, tk,…., tn and the extinction time of the whole population will be the maximum of this set:  . The cumulative distribution function of the maximum time
. The cumulative distribution function of the maximum time  will be the probability of the joint event that each sub-population k has an extinction time less than
will be the probability of the joint event that each sub-population k has an extinction time less than  :
:
 where P(t) is the CDF for a single population given by Eqn.10 in the main text, but with I0 → I0/n. Given the form of Eqn.10, these calculations can be performed exactly, whereas using extreme value theory it usually required that the tails of the distribution asymptotically obey some exponential form, which allows approximate calculation. Doing these calculations we find
where P(t) is the CDF for a single population given by Eqn.10 in the main text, but with I0 → I0/n. Given the form of Eqn.10, these calculations can be performed exactly, whereas using extreme value theory it usually required that the tails of the distribution asymptotically obey some exponential form, which allows approximate calculation. Doing these calculations we find  , where
, where  . It is then simple to show that the n-dependence cancels in the final result to give
. It is then simple to show that the n-dependence cancels in the final result to give

In other words, population sub-division into equal sized isolated populations does not affect the extinction time distribution of the whole global population. In fact, it is simple to extend these arguments to any population sub-division, where  , where Ik is the initial infected population in each, as long as the fraction of susceptible and Re is the same in each sub-population. This is not surprising, as it is just a restatement of the mean-field/well-mixed approximation that infected individuals and sub-populations all experience the same probability of encountering a susceptible individual S0/N which is set by the global number of susceptible individuals S0.
, where Ik is the initial infected population in each, as long as the fraction of susceptible and Re is the same in each sub-population. This is not surprising, as it is just a restatement of the mean-field/well-mixed approximation that infected individuals and sub-populations all experience the same probability of encountering a susceptible individual S0/N which is set by the global number of susceptible individuals S0.
Footnotes
a Electronic mail: bkhatri{at}imperial.ac.uk
REFERENCES




Subject Area
Reviews and Context
0
Comment
0
TRIP Peer Reviews
0
Community Reviews
0
Automated Services
2
Blogs/Media
Author Videos