
Abstract
Introduction Autoimmune diseases are heterogeneous and often lack specific or sensitive diagnostic tests. Increased percentages of CD4+CXCR5+PD1+ circulating T follicular helper (cTfh) cells and skewed distributions of cTfh subtypes have been associated with autoimmunity. However, cTfh cell percentages can normalize with immunomodulatory treatment despite persistent disease activity, indicating the need for identifying additional cellular and/or serologic features correlating with autoimmunity.
Methods The cohort included 50 controls and 56 patients with autoimmune cytopenias, gastrointestinal, pulmonary, and/or neurologic autoimmune disease. Flow cytometry was used to measure CD4+CXCR5+ T cell subsets expressing the chemokine receptors CXCR3 and/or CCR6: CXCR3+CCR6− Type 1, CXCR3−CCR6− Type 2, CXCR3+CCR6+ Type 1/17, and CXCR3− CCR6+ Type 17 T cells. IgG and IgA autoantibodies were quantified using a microarray featuring 1616 full-length, conformationally intact protein antigens. The 97.5th percentile in the control cohort defined normal limits for T cell subset percentages and total number (burden) of autoantibodies.
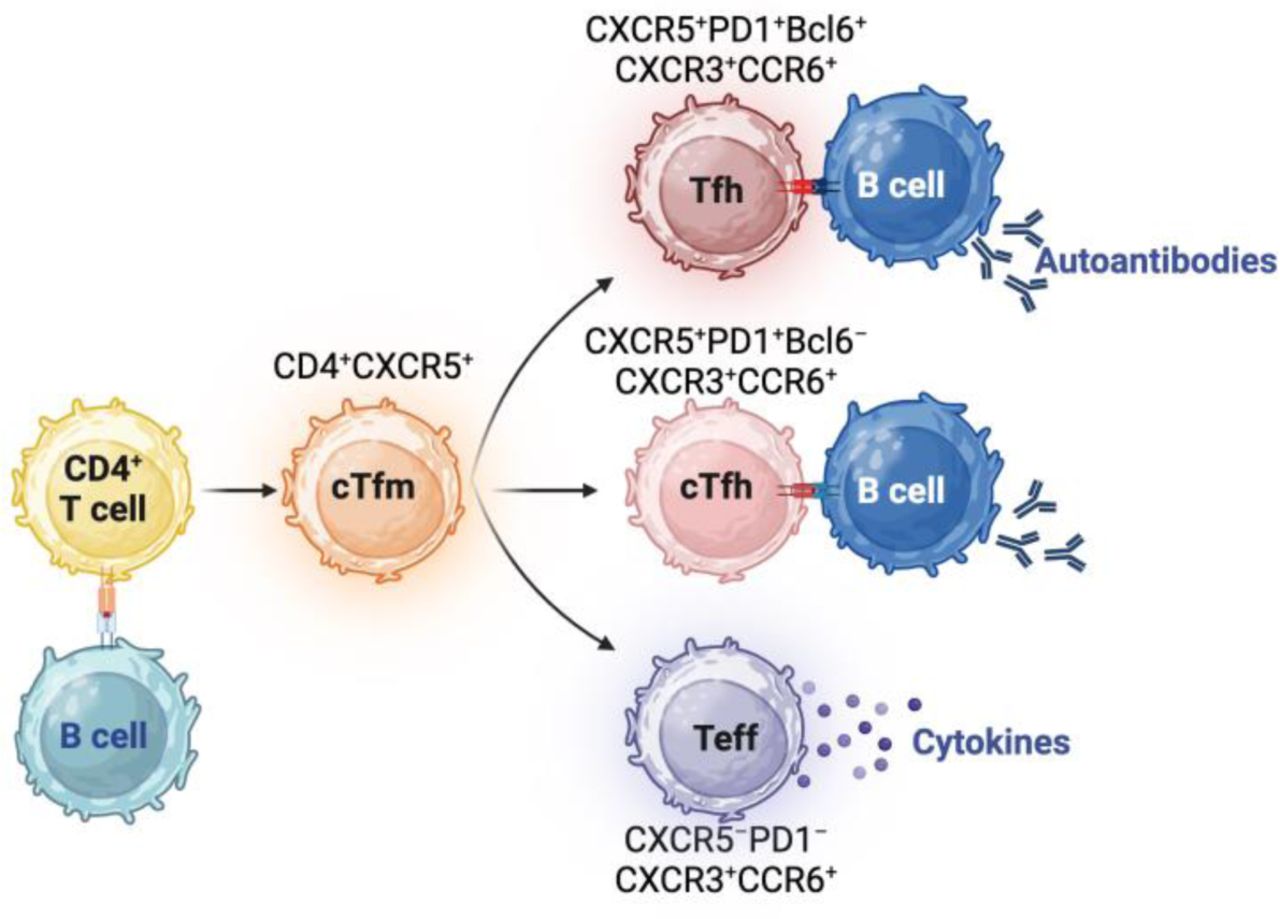
Results This study focused on CD4+CXCR5+ T cells because CXCR5 upregulation occurs after cognate T-B cell interactions characteristic of autoimmune diseases. We refer to these cells as circulating T follicular memory (cTfm) cells to acknowledge the dynamic nature of antigen-experienced CXCR5+ T cells, which encompass progenitors of cTfh or Tfh cells as well as early effector memory T cells that have not yet lost CXCR5. Compared to controls, 57.1% of patients had increased CXCR5+CXCR3+CCR6+ cTfm1/17 and 25% had increased CXCR5+CXCR3−CCR6+ cTfm17 cell percentages. Patients had significantly more diverse IgG and IgA autoantibodies than controls and 44.6% had an increased burden of autoantibodies of either isotype. Unsupervised autoantibody clustering identified three clusters of patients with IgG autoantibody profiles distinct from those of controls, enriched for patients with active autoimmunity and monogenic diseases. An increased percentage of cTfm17 cells was most closely associated with an increased burden of high-titer IgG and IgA autoantibodies. A composite measure integrating increased cTfm1/17, cTfm17, and high-titer IgG and/or IgA autoantibodies had 91.1% sensitivity and 90.9% specificity for identifying patients with autoimmunity. Percentages of cTfm1/17 and cTfm17 percentages and numbers of high-titer autoantibodies in patients receiving immunomodulatory treatment did not differ from those in untreated patients, thus suggesting that measurements of cTfm can complement measurements of other cellular markers affected by treatment.
Conclusions This study highlights two new approaches for assessing autoimmunity: measuring CD4+CXCR5+ cTfm subsets as well as total burden of autoantibodies. Our findings suggest that these approaches are particularly relevant to patients with rare autoimmune disorders for whom target antigens and prognosis are often unknown.

Introduction
The clinical heterogeneity of autoimmune diseases complicates diagnosis of these disorders. Due to limitations in diagnostic testing, some remain diagnoses of exclusion, such as immune thrombocytopenia (ITP), autoimmune neutropenia or seronegative autoimmune hepatitis.[1,2] Positivity for disease-associated autoantibodies does not consistently correlate with disease activity due to the variable recognition of epitopes inherent in a polyclonal response. Furthermore, many disease-associated autoantibodies recognize intracellular antigens whose contribution to disease remains unknown; even more disorders have no known autoantibodies.[3–5] Although next-generation DNA sequencing has identified some monogenic causes of autoimmunity, most patients lacks a genetic diagnosis.[6] Even relatively common autoimmune disorders with defined clinical criteria and disease-associated autoantibodies, such as systemic lupus erythematosus (SLE) and multiple sclerosis, can have diagnostic delays upwards of three years.[7,8] Delayed diagnoses contribute to disease progression and morbidity through postponed or inadequate treatment,[9–11] highlighting the need for additional approaches to reduce diagnostic delay.
Autoimmunity arises from self-reactive T and B cells that generate inflammatory cytokines and autoantibodies.[12] T follicular helper (Tfh) cells are CD4+ T cells with robust capacity for inducing B cell activation and class-switching.[12] Upon recognizing antigen, T cells upregulate the chemokine receptor CXCR5.[13] While effector T cells lose expression of CXCR5 after terminal differentiation, Tfh cell progenitors maintain expression of CXCR5, which enables their migration to the interface of the T cell zone and B cell follicles in lymphoid organs.[12] Subsequently, sustained interactions between Tfh and B cells upregulates markers characteristic of Tfh cells in germinal centers: the transcription factor B cell lymphoma 6 protein (Bcl6) and the immune checkpoint receptor, programmed cell death 1 (PD-1).[12,14] Notably, the periphery contains CD4+CXCR5+PD1+ T cells that lack Bcl6.[15,16] These cells are commonly referred to as circulating Tfh (cTfh) cells due to features shared with Tfh cells in the germinal center, including T cell receptor clonotypes and the ability to induce class-switching.[15–18] We and others have previously shown expansion of CD4+CXCR5+PD1+ cTfh cells in patients with multi-systemic autoimmune disorders, such as activated protein kinase delta syndrome and SLE.[12,19] However, increased percentages of cTfh cells are not a universal feature of all autoimmune disorders and can normalize with immunosuppressive treatment even in patients with persistent disease activity,[20] indicating the need for additional measures of autoimmunity.
Like effector T cells, which include Th1, Th2, Th17, and Th1/17 subsets, cTfh cells can be divided into subtypes based on expression of the chemokine receptors CXCR3 and CCR6.[20–22] Commonly expanded during viral and parasitic infections or after vaccination, cTfh1 cells robustly produce IFN-γ and can activate memory, but not naïve, B cells.[22–25] Compared to cTfh1 cells, cTfh2 and cTfh17 cells are considered more potent activators of B cells because they enable naïve B cell differentiation to class-switched plasmablasts, with cTfh17 cells enhancing IgA and IgG secretion and cTfh2 cells enhancing IgE and IgG secretion.[15] IL-4 promotes the differentiation of CXCR3−CCR6− cTfh2 cells, which produce IL-4 and IL-5.[15] TGF-β1 and IL-6 enable the differentiation of CXCR3−CCR6+ cTfh17 cells.[15] Increased perceentages of cTfh17 and cTfh2 cells have been found in patients with autoimmunity, including SLE, psoriasis, Sjogren’s syndrome, myasthenia gravis, multiple sclerosis, IgG4-related disease, and juvenile dermatomyositis.[15,26,27] Additionally, expansions of cTfh subsets occurring in healthy individuals during infections are thought to contribute to the production of non-pathogenic autoantibodies in the general population.[28] CXCR3+CCR6+ cTfh1/17 cells share features of cTfh1 and cTfh17 cells, such as dual production of IFN-γ and IL-17,[29] but much less is known about the associations between cTfh1/17 cells and autoimmunity.
Given the essential contributions of T and B cells to the development of autoimmune disorders, we hypothesized that aberrations in T cell subsets and autoantibody repertoire could reflect germinal center dysfunction, and identify autoimmunity even in patients with normal percentages of total CXCR5+PD1+ cTfh cells. To complement prior studies investigating subset skewing among CD4+CXCR5+PD1+cTfh cell subsets in patients with autoimmunity,[16,30] we focused on a larger population of CD4+CXCR5+ T cells since CXCR5 upregulation occurs after antigen-dependent, cognate interactions[13] essential to the pathogenesis of autoimmunity. CD4+CXCR5+ T cells include PD-1− effector T cells that have not yet downregulated CXCR5, early cTfh cells with low to intermediate PD-1 expression, and cTfh cells near terminal differentiation with robust PD-1 expression – all of which can promote the generation of proinflammatory cytokines and autoantibodies characteristic of autoimmune disorders.
Methods
2.1 Study participants
Informed consent was obtained from patients, all of whom were recruited at Boston Children’s Hospital. This study was approved by the Institutional Review Board of Boston Children’s Hospital.
Information regarding medical diagnoses and treatments received was obtained through review of the electronic medical record. Race and ethnicity were obtained from self-reported data from the medical record. The control cohort included patients without any history of autoimmunity, immune dysfunction, malignancy, or transplantation. Patients were classified as having a diagnosis of autoimmunity based on clinical, laboratory, or genetic causes abstracted from the medical record by the study’s clinical hematologists and immunologists prior to measurement of cTfh cells to minimize investigator bias. Disease activity was defined by abnormal laboratory values consistent with the respective autoimmune diagnosis or autoimmune disease-specific symptoms or cytopenias requiring medications. Intravenous immunoglobulin (IVIG) given within 4 weeks of enrollment or rituximab given within 12 months of enrollment was considered ongoing active treatment based on published kinetics of B cell repopulation in patients with autoimmune disorders.[31,32] For patients with autoimmune hemolytic anemia (AIHA), rituximab response was defined as >2 g/dL increase in hemoglobin from time of diagnosis or normalization of hemoglobin with or without biochemical resolution of hemolysis.[33] For patients with ITP, rituximab response was defined as platelet count ≥30 × 109/L and at least 2-fold increase of the baseline count within 3 months of first dose.[34] One patient received rituximab for interstitial lung disease; response was defined by improvement on pulmonary function tests.
2.2 Immunophenotyping
To minimize T cell death that can occur during the process of cell isolation,[35] flow cytometric assessment of T cells was done on 100 µL of whole blood collected in sodium heparin anticoagulant tubes. Cell surface staining was performed using the following antibodies: CD4 (BioLegend #317420), PD-1 (BioLegend, #329907), CXCR5 (BioLegend, #356904), CXCR3 (BioLegend, #353716), and CCR6 (BioLegend, #353434). Similar percentages of cTfm subsets were obtained when anti-CD45RA (BioLegend #304129) was added to exclude naïve T cells. Flow cytometry data were acquired on BD LSRFortessa and were analyzed using FlowJo™ v10.8 Software (BD Life Sciences). The upper limit of normal was defined as the 97.5th percentile of control values for T cell phenotyping, similar to that of other clinical laboratory tests.[36,37] Increased percentages of CD4+CXCR5+PD-1+ cTfh cells was defined using a threshold of 12% as previously published.[19]
2.3 Autoantibody detection
Sengenics KREX technology (i-OME Discovery array in combination with dual color IgG/IgA detection) was used to profile seroreactivity against 1616 self-antigens with preserved native conformation.[38,39] Controls for this platform included 22 individuals from the above described control cohort as well as a commercially available pooled normal plasma (Sigma) from 50 – 70 individuals without clinical evidence of autoimmunity. Plasma was diluted 1:200 for the assay. Autoantibody binding to antigens was measured using an open format microarray laser scanner (Agilent) at 10 µm resolution, with quantification of intensities using GenePix Pro 7 software (Molecular Devices). Net fluorescence intensities were calculated by subtracting background signaling intensities, followed by log2 transformation, Loess normalization[40] using the normalizeCyclicLoess() function[41] from R package limma[42]. Batch-correction was performed using the ComBat() function[43] from R package sva.[44] To avoid potential confounding from intravenous immunoglobulin (IVIG), we excluded autoantibodies with significantly increased intensities in patients who received IVIG within 4 weeks of study enrollment compared to patients who did not receive IVIG.
2.4 Statistical analysis
Differences in the Shannon diversity indices [45] were calculated using Hutcheson’s t test.[46] Unsupervised hierarchical clustering was performed using Euclidean distance and the Ward D2 method with the ComplexHeatmap R package.[47,48] Sensitivity and specificity of the composite measure for autoimmunity were calculated using Fisher’s exact test. Statistical tests were performed using Prism v10.0.0 (GraphPad) or R version 4.3.1. Graphs were created with Prism v10.0.0 and illustrations were created with BioRender.com.
Results
3.1 Patient characteristics
This study enrolled 106 individuals: 56 patients with autoimmune diseases and 50 controls (Table 1). The heterogeneity of this patient cohort reflects the phenotypic variability in clinical practice. Immune cytopenias were the most common clinical feature, affecting 84% of the patients. This included 11 patients with ITP, 15 with AIHA, and 22 with Evans syndrome. Additional outcomes of autoimmunity in this cohort included hepatitis, intestinal disease, interstitial lung disease, neurologic disease, and thyroiditis (Table 1). There were 17 patients (30.3%) with a monogenic diagnosis (Supplementary table 1). At enrollment, 50 patients (89.3%) had active disease and 40 (71%) were receiving treatment (Table 1). A total of 16 patients had a history of rituximab treatment prior to enrollment, of whom eight had received it within 12 months before enrollment. Among the 16 patients who received rituximab, 11 (68.8%) had quantitative evidence of response to rituximab, as detailed in the methods.
3.2 Patients with autoimmunity have expanded cTfm1/17 and cTfm17 subsets
We have previously shown that a percentage of total CD4+CXCR5+PD-1+ cTfh cells exceeding 12% of CD4+ T cells has 88% sensitivity and 94% specificity for active, multi-systemic autoimmunity, and can normalize with immunomodulatory treatment.[19] In contrast to our prior study, the majority of this cohort (69.6%, 39 patients) had a normal percentage of total CD4+CXCR5+PD-1+ cTfh cells: six had inactive disease, four had immune cytopenias without evidence of multi-systemic disease, and the remaining 28 were on immunomodulatory therapies (Fig. 1A). In this study, we focused on CXCR5+ expressing CD4+ since robust and rapid upregulation of this chemokine receptor occurs primarily after cognate interactions with antigen-presenting cells, rather than non-specific T cell activation with anti-CD3/CD28 stimulation or PHA.[13] As CD4+CXCR5+ T cells with variable PD-1 expression share features of CXCR5+PD1+ cTfh cells, CXCR3+ memory Th1 cells, and CCR6+ memory Th17 cells, we refer to them as circulating T follicular memory (cTfm) cells. To characterize CD4+CXCR5+ cTfm cell subsets, cTflm1, cTfm2, cTfm17, and cTfm1/17 cells were identified based on the expression of CXCR3 and CCR6 (Fig. 1B). These subsets include PD-1+ cTfm cells as well as PD-1− cTfm cells. CD4+CXCR5+PD1− cells can secrete pro-inflammatory cytokines and develop into PD-1+ cells.[49–51] Using the 97.5th percentile of cTfm subset percentages in controls as the upper limit of normal, 32 (57.1%) patients had increased cTfm1/17 percentages, and all but one had active disease (Fig. 1C). Increased cTfm17 percentages were found in 14 (25%) patients, of whom two had inactive disease (Fig. 1C). cTfm1 and cTfm2 subsets were comparable between the patients and controls (Fig. 1C). Within the cohort with autoimmunity, percentages of cTfm subsets in patients who were on treatment did not differ from those in patients not on treatment (Fig. 1D).

A. Percentages of total CD4+CXCR5+PD-1+ Tfh cells in patients and controls. Dotted line at 12% represents the upper limit of normal, as previously published.[19] B. Gating strategy for identifying cTfm cell subsets. C. Percentages of cTfm1, cTfm1/17, cTfm17, and cTfm2 cells in patients and controls. The dotted line represents the 97.5% in controls. D. Percentages of cTfm cell subsets in patients who were either treated or untreated at the time of enrollment.
3.3 Autoantibody results
We next investigated autoantibody profiles in our autoimmunity cohort of 56 patients, along with 22 individuals from the previously described control group. We investigated profiles of IgG and IgA autoantibodies using a platform that detects IgG and IgA autoantibodies to 1616 conformationally intact, full-length human proteins related to autoimmunity and cancer.[38,39] Due to the heterogeneity of clinical features associated with immune cytopenias,[1,52,53] this cohort was not powered to identify enrichment of specific autoantibodies for clinical phenotypes. We instead hypothesized that patients with autoimmunity would have either an increased number and/or diversity of autoantibodies compared to controls. High-titer autoantibodies were classified as those with normalized intensities exceeding three standard deviations of those found in controls. This approach identified a high-titer anti-muscle-specific tyrosine kinase in a patient with myasthenia gravis in the setting of Kabuki syndrome, concordant with clinical testing for autoantibodies in this patient.
Patients had a median of five high-titer IgG autoantibodies (range: 0 – 249 autoantibodies per patient) compared to a median of 1.5 high-titer IgG autoantibodies in controls (range: 0 – 16 autoantibodies per control, Fig. 2A). High-titer IgG autoantibodies in patients with autoimmunity were significantly more diverse than those in controls, as measured by the Shannon diversity index (Fig. 2B). Additionally, there were 521 high-titer IgG autoantibodies found only in the patients with autoimmunity (Supplementary data 1), whose targets were enriched in pathways pertaining to cell cycling, apoptosis, signaling downstream of GATA2, a transcription factor essential for hematopoietic cell development,[54,55] and PI3K/AKT/mTOR, a metabolic pathway important for cTfh and effector T cell dysregulation as well as autoimmune cytopenias [56] (Fig. 2C). Among the nine patients with active disease who were not on immunomodulatory treatment at the time of enrollment, eight had a total number of high-titer IgG autoantibodies comparable to those in controls, suggesting persistent cellular, rather than serologic, autoimmunity after prior therapy.

A. Total number of high-titer IgG autoantibodies. B. Diversity of high-titer IgG autoantibodies, as measured by the Shannon diversity index. C. Pathways enriched in high-titer IgG autoantibodies found only in the patient cohort. D. Total number of high-titer IgA autoantibodies. E. Diversity of high-titer IgA autoantibodies, as measured by the Shannon diversity index. F. Pathways enriched in high-titer IgA autoantibodies found only in the patient cohort. ****p<0.0001
Quantification of IgA autoantibodies identified a median of two high-titer IgA autoantibodies (range: 0 – 232) and increased autoantibody diversity in patients, compared to a median of one high-titer IgA autoantibody in controls (range: 0 – 5, Fig. 2D, E). There were 523 high-titer IgA autoantibodies found only in the patients with autoimmunity (Supplementary Data 2), whose targets were significantly enriched in pathways pertaining to apoptosis, cellular senescence, cell cycling, and cytokine signaling (Fig. 2F). These pathways have been previously shown to contribute to autoimmunity and overlap with those enriched in targets of high-titer IgG autoantibodies.[57,58] In this cross-sectional study, the number of high-titer IgG or IgA autoantibodies was not statistically correlated with differences in disease activity or treatment (Supplementary Fig. 1). The targets of high-titer IgG and IgA autoantibodies were predominantly intracellular antigens, likely reflecting antigen release after cell death.[59] To complement this analysis of high-titer autoantibodies, we performed unsupervised clustering of all autoantibodies in patients and controls. There were four clusters collectively composed of 21 patients with IgG autoantibody profiles distinct from those of controls (clusters 2, 3, 6, and 7 in Supplementary Fig. 2). These clusters were enriched for patients with active autoimmunity (n=19, 90.5%) and monogenic diseases (n=12, 63.2%). Among the patients lacking a definitive genetic diagnosis, three have candidate variants under ongoing investigation. One control (cluster 1) is a two year old boy who started daycare approximately four months prior to study enrollment. He had a normal immunologic evaluation and a history of mild upper respiratory infections that resolved after six months in daycare without any clinical evidence of autoimmunity. His high titer IgG autoantibody pattern is likely due to his history of recurrent upper respiratory infections upon daycare entry, thereby separating him from the patients and other controls. In contrast, unsupervised clustering of IgA autoantibody profiles identified only four patients with autoantibody profiles distinct from controls, all of whom had active AIHA (n=3) or ITP (n=1) (Supplementary Fig. 3). Collectively, these findings show that a broad autoantibody screen can identify patients with a large burden of high-titer, diverse autoantibodies, even for disorders lacking identifiable shared or pathogenic autoantibodies.
3.4 Combining cTfh subsets with autoantibody profiling in the diagnosis of autoimmunity
Based on the dual contributions of T and B cells in the development of autoimmunity, we tested the sensitivity and specificity of a composite measure integrating cTfm1/17, cTfm17, and increased burden of high-titer IgG and/or IgA autoantibodies for autoimmunity. To create this composite measure, we tabulated the number of laboratory features exceeding the 97.5th percentile of controls as follows: (1) cTfm1/17 percentage >36.0%, (2) cTfm17 percentage >40.1%, (3) >14 high-titer IgG autoantibodies, and (4) >4 high-titer IgA autoantibodies (Table 2). Within the control cohort, most (n=20, 90.9%) had a composite measure of 0, while two had a composite measure of 1 due to an increased burden of high-titer IgG or IgA autoantibodies. The patients with autoimmunity had composite measures of zero (n=5, 8.9%), one (n=34, 60.7%), two (n=14, 25%), or three (n=3, 5.4%) as summarized in Table 2 and Fig. 3A. Of the five patients with a composite measure of zero, four had active ITP and one had AIHA in remission. A composite measure >1 had a sensitivity of 91.1% and a specificity of 90.9% for autoimmune disease. Unsupervised clustering of the variables included in the composite measure revealed that an increased percentage of cTfm1/17 cells was most closely associated with a positive composite measure (Fig. 3B). An increased percentage of cTfm17 cells was most closely associated with an increased burden of high-titer IgG and IgA autoantibodies (Fig. 3B), consistent with the known contribution of cTfm17 cells to both autoantibody isotypes.[29,60]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
A. Composite measures in controls and patients, with indicated clinical phenotypes and genetic diagnoses. AIHA, autoimmune hemolytic anemia; ITP, immune thrombocytopenia; ES, Evans Syndrome; GOF, gain of function. B. Unsupervised clustering of composite measure components and patients.
Individuals with no autoimmunity included 20 controls and 2 patients having Kabuki syndrome without autoimmunity.
We conclude with an illustrative example of how this composite measure can be used for disorders with variably penetrant autoimmunity. Our cohort includes six patients with Kabuki Syndrome due to pathogenic variants in KMT2D (Supplementary table 1), which encodes a methyltransferase that regulates the expression of genes contributing to a diverse range of cellular functions.[61] The clinical phenotype of Kabuki Syndrome is unpredictable since some, but not all patients develop immune dysfunction that may include hypogammaglobulinemia and/or autoimmunity, such as vitiligo, autoimmune thyroiditis, myasthenia gravis, and immune cytopenias.[61] Of the six patients with Kabuki syndrome in our cohort, four had clinically detectable autoimmunity. Three of these had a positive composite measure due to increased cTfm1/17 percentages, cTfm17 percentages, and/or burden of high-titer IgG autoantibodies. Two had an increased percentage of total cTfm cells exceeding the normal threshold of 12%. The remaining two patients lacking autoimmunity had a composite measure of zero. These findings suggest that combined assessments of cTfh cells and high-titer autoantibodies may be particularly relevant for patients with disorders characterized by variable autoimmunity.
Discussion
Many clinically heterogenous autoimmune disorders are diagnoses of exclusion due to limited targeted diagnostic testing, and are managed with a “watch and wait” approach that can delay initiation of effective treatment.[62] The term cTfm acknowledges the dynamic nature of antigen-experienced CXCR5+ T cells, which can differentiate into peripheral cTfh cells or germinal center Tfh cells with continued expression of CXCR5+, or downregulate CXCR5+ as they differentiate into effector memory T cells. Here, we demonstrate the utility of integrating CD4+CXCR5+ cTfm subsets and serologic markers for identifying autoimmunity, even in patients undergoing immunomodulatory treatment. We show that patients with autoimmunity have skewed cTfm subsets, with increased percentages of cTfm1/17 and cTfm17 cells. Using broad autoantibody profiling, we identify an increased burden of high-titer IgG or IgA autoantibodies in 37.5% of the patients studied, thereby providing another approach for assessing serologic autoimmunity when known pathogenic autoantibodies are unknown or undetectable.
We have previously shown that circulating CD4+CXCR5+PD1+ cTfh cells exceeding 12% of CD4+ T cells have high sensitivity and specificity for active, multi-system autoimmunity,[19] complementing published studies showing increased cTfh levels in patients with SLE, rheumatoid arthritis, Sjogren syndrome, and autoimmune neurologic disorders.[12,20] Less is known about the dysregulation of cTfh cells in patients with autoimmune cytopenias, which affected 84% of the patients studied. In a cohort of 24 patients with Evans Syndrome and 22 with chronic ITP, Kumar et al. found that patients with Evans Syndrome have increased percentages of total cTfh and cTfh1 cells, with a concomitant reduction in cTfh17 cells compared to those with chronic ITP.[30] Notably, this study’s evaluation of cTfh subsets focused on PD1+ cells.[30] This is distinct from our approach of measuring CD4+CXCR5+ T cells that are either PD1− or PD1+. PD-1 serves as a rheostat that tightly regulates the development of Tfh cells in the germinal center. Upon binding PD-L1 on B cells, PD-1 promotes class-switching and germinal center development by enhancing the secretion of IL-21.[63] PD-1 also restrains Tfh cell development by reducing T cell activation.[64] Furthermore, PD-1 restricts the egress of Tfh cells from germinal centers by limiting expression of CXCR3, a chemokine receptor that binds to ligands expressed by dendritic cells outside the lymphoid follicles and peripheral stromal cells, such as endothelial cells, fibroblasts, and keratinocytes.[64] High expression of PD-1 thus enables precise control of T cell activation, migration, and function.[64] In contrast, CD4+CXCR5+ T cells with low PD-1 expression have been found to have multiple fates and functions. CD4+CXCR5+CXCR3+PD1− cTfh cells can differentiate into PD1+ cTfh cells.[49,50] Additionally, CXCR5+CCR6+PD1−cells produce greater amounts of proinflammatory cytokines, such IL-17 and TNF-α.[51] Inclusion of both PD1+ and PD1− CXCR5+ cTfm cells thus allows us to include different types of antigen-experienced T cell populations with distinct contributions to autoimmunity.
We found increased percentages of cTfm17 and cTfm1/17 in patients with diverse types of autoimmunity, even in patients receiving immunosuppressive therapies. Effector T cells expressing CXCR3 and CCR6 have been shown to resist glucocorticoid-induced cell death due to the expression of the multi-drug resistance type 1 membrane transporter.[65] Additional studies are needed to determine cTfm cells similarly express drug resistance transporters. Unsupervised clustering indicated that increased percentages of cTfm17 cells, but not cTfm1/17 cells, were associated with high titers of IgG and IgA autoantibodies in our cohort. Expanded CD4+CXCR5+PD1+CCR6+ cTfh17 cells has been found in patients with a range of autoimmune disorders, including SLE, systemic sclerosis, myasthenia gravis, and Hashimoto’s thyroiditis, and can positively correlate with pathogenic autoantibodies.[16,20,66,67] The associations of cTfh cells with dual expression of CXCR3 and CCR6 with autoimmune disease are less well-known, although recent studies have identified increased percentages of these cells in patients with rheumatoid arthritis and multiple sclerosis [68,69]. More is known about Th1/17 cells, the effector cell counterpart of cTfh1/17 cells also enriched in patients with autoimmunity.[70] Effector Th1/17 cells are multifunctional, with the capacity to secrete proinflammatory cytokines.[71–73] Further investigations are needed to determine the mechanisms underlying the expansion of cTfm1/17 cells in patients with autoimmunity and if these cells exhibit polyfunctionality similar to that of effector Th1/17 cells.
We complemented these studies of cTfm subsets with a broad autoantibody profiling to assess the overall burden of high-titer autoantibodies since disease-associated autoantibodies are unknown for many autoimmune disorders. Even for disorders with known autoantibodies, the relationship between autoantibodies and disease activity is unpredictable, since autoantibodies can develop before clinical manifestations of autoimmune diseases.[62] Autoantibodies can also exist in healthy individuals, arising from molecular mimicry with infectious pathogens and natural loss of self-tolerance with aging, among other factors.[28,74,75] Most disease-associated autoantibodies are of the IgG isotype, followed by IgA autoantibodies.[76] Our patient cohort with autoimmunity harbored a diverse set of high-titer IgG and IgA autoantibodies not found in controls, with more patients having an increased number of high-titer IgA than IgG autoantibodies. The high-titer IgA autoantibodies unique to the patients targeted self-antigens enriched in pathways similar to those targeted by the IgG autoantibodies, suggesting that screening for IgA autoantibodies may comprise another readout for some patients with autoimmunity. Although the autoantibody diversity in our patient cohort may be partly attributed to the heterogeneous clinical phenotypes in our cohort, published studies have similarly identified broad autoantibody repertoires in patients with SLE and in the elderly.[77,78] This contrasts with the “antibodyomes” of healthy individuals sharing common autoantibodies,[28] which eventually become more diverse as self-tolerance diminishes with aging.[78–80] The presence of anti-cytokine autoantibodies in patients is thought to be a proxy for loss of tolerance as they have not consistently been shown to be pathogenic or disease-defining in patients with autoimmunity.[81] They have subsequently been described in autoimmune conditions, such as SLE, rheumatoid arthritis, and multiple sclerosis. In our patient cohort, we identified high-titer anti-IL1A (n=1) and anti-IL24 (n=2) IgG autoantibodies. The autoantibody platform used in our study does not quantify all known anti-cytokine autoantibodies.
In combining measurements of cTfm subsets with the burden of high-titer IgG and IgA autoantibodies, we developed a composite measure with high sensitivity and specificity for autoimmunity. The diversity of clinical phenotype, disease activity, and therapies in our patient cohort reflects the reality of clinical practice. It is challenging to determine whether a cytopenia has an autoimmune etiology. For example, the differential diagnosis for isolated thrombocytopenia includes ITP as well as many other non-autoimmune causes, including familial thrombocytopenia, bone marrow failure syndromes, medication-induced thrombocytopenia, and platelet sequestration in the liver or spleen.[82] ITP remains a diagnosis of exclusion, as there are no laboratory tests for diagnosing ITP and current clinical tests for anti-platelet glycoprotein autoantibodies have 53% sensitivity for ITP.[82] Even at centers with a large volume of immune cytopenia cases, up to 14% of children and 12.2% of adults who were diagnosed with ITP were misdiagnosed, while 3.1% of patients diagnosed with other causes of thrombocytopenia were ultimately found to have ITP.[83,84] Similarly, autoimmune neutropenia is a diagnostic challenge due to the lack of a diagnostic tests specific for autoimmune neutropenia.[52] Children with autoimmune neutropenia typically have excellent outcomes with relatively few infectious complications. In contrast, those with neutropenia due to impaired bone marrow production can have significant infectious risks.[52] Notably, we were able to detect increased percentages of cTfm17 and cTfm1/17 cells even in patients with ongoing immunomodulatory treatment. Therefore, a composite measure based on cellular and humoral mechanisms of autoimmunity, rather than disease-specific antigens, may address these diagnostic challenges.
This study has several limitations. An independent validation cohort is needed to confirm the sensitivity and specificity of cTfm subset skewing and high-titer autoantibodies for autoimmunity. In this cross-sectional study, we did not evaluate longitudinal changes in cTfh profiles and total autoantibody burden relative to changes in disease progression or clinical responses to treatment. Only one prior study has measured changes in the total autoantibody burden after treatment, in which a clinical response to rituximab was associated with a reduced number of autoantibodies detected by an array measuring 1,292 IgG, IgA, and IgD autoantibodies in a cohort of 26 patients with rheumatoid arthritis.[85] Future studies are needed to identify functional differences among skewed cTfh cell subsets that correlate with disease mechanisms, phenotypes, and activity. Our findings are not intended to establish a clinical tool for the diagnosis of autoimmunity but rather to demonstrate that integration fo these components are distinguishing features of autoimmunity.
Here, we have applied two consequences of sustained T-B cell crosstalk – expansion of CD4+CXCR5+ cTfm cell subsets and autoantibodies – for the diagnosis of patients undergoing treatment for diverse types of autoimmunity. In our cohort, cTfm1/17 cells were the most commonly expanded subset, thus identifying a potential role for these cells in the pathogenesis of immune cytopenias. We show that broad autoantibody screening can be used to identify patients with large burdens of autoantibodies as an additional proxy for autoimmunity in patients lacking disease-associated antibodies.
Data Availability
Research data will be made available upon request to the authors
Supplementary Figure 1. A) Number of high-titer IgG and IgA autoantibodies in patients with active (+) and inactive (−) disease. B) Number of high-titer IgG and IgA autoantibodies in patients on treatment (+) and not on treatment (−) at time of enrollment
Supplementary Figure 2. Heatmap with unsupervised clustering based on the normalized net intensities of IgG autoantibodies in patients and controls. The STAT1, STAT2, and STAT3 variants are all gain of function variants. Red = presence, Grey = inactive, White = not present (control).
Supplementary Figure 3. Heatmap with unsupervised clustering for IgA. Red = presence, Grey = inactive, White = not present (control)
Supplementary Table 1: Genetic diagnoses of patients with monogenic disorders
Supplementary Data 1. High titer IgG autoantibodies with subcellular locations
Supplementary Data 3: High titer IgA autoantibodies with subcellular locations
Conflicts of Interest
EMH, SC, LS, SS, CY, AN, MG-A, CDP, BL, JC have no conflicts of interest to disclose. DHK is a consultant for Adivo Associates. RFG receives research funding from Novartis, Sobi, and Agios and is a consultant for Agios and Sanofi. JP is an employee of Sengenics. LAH received salary support from the Childhood Arthritis and Rheumatology Research Alliance, investigator-initiated research grants from Bristol Myers Squibb, and consulting fees from Sobi.
Funding
This work was supported by the National Institutes of Health (T32HL007574 to EMH, T32AI007512 to SC, BL, JC, and R01DK130465 to JC) and the Immune Deficiency Foundation (BL). The Children’s Rare Disease Cohort initiative[86] funded the whole exome sequencing of study participants. Sources of funding had no role in the study design or execution, data interpretation, or writing of the manuscript.
Acknowledgments
We are grateful to our patients and their families for their participation in this study.
Footnotes
Tables included, Figures 1 and 3 revised
Abbreviations
- AIHA
- Autoimmune hemolytic anemia
- cTfh
- Circulating T follicular helper cell
- cTfm
- Circulating T follicular memory cell
- ES
- Evans syndrome
- ITP
- Immune thrombocytopenia
- IVIG
- Intravenous immunoglobulin
- SLE
- Systemic lupus erythematosis
References
- [1].↵
- [2].↵
- [3].↵
- [4].
- [5].↵
- [6].↵
- [7].↵
- [8].↵
- [9].↵
- [10].
- [11].↵
- [12].↵
- [13].↵
- [14].↵
- [15].↵
- [16].↵
- [17].
- [18].↵
- [19].↵
- [20].↵
- [21].
- [22].↵
- [23].
- [24].
- [25].↵
- [26].↵
- [27].↵
- [28].↵
- [29].↵
- [30].↵
- [31].↵
- [32].↵
- [33].↵
- [34].↵
- [35].↵
- [36].↵
- [37].↵
- [38].↵
- [39].↵
- [40].↵
- [41].↵
- [42].↵
- [43].↵
- [44].↵
- [45].↵
- [46].↵
- [47].↵
- [48].↵
- [49].↵
- [50].↵
- [51].↵
- [52].↵
- [53].↵
- [54].↵
- [55].↵
- [56].↵
- [57].↵
- [58].↵
- [59].↵
- [60].↵
- [61].↵
- [62].↵
- [63].↵
- [64].↵
- [65].↵
- [66].↵
- [67].↵
- [68].↵
- [69].↵
- [70].↵
- [71].↵
- [72].
- [73].↵
- [74].↵
- [75].↵
- [76].↵
- [77].↵
- [78].↵
- [79].
- [80].↵
- [81].↵
- [82].↵
- [83].↵
- [84].↵
- [85].↵
- [86].↵



