
Abstract
Aim Variants in the SLCO1B1 (solute carrier organic anion transporter family member 1B1) gene encoding the OATP1B1 (organic anion transporting polypeptide 1B1) protein are associated with altered transporter function that can predispose patients to adverse drug effects with statin treatment. We explored the effect of six rare SLCO1B1 single nucleotide variants (SNVs) occurring in Finnish individuals with a psychotic disorder on expression and functionality of the OATP1B1 protein.
Methods The SUPER-Finland study has performed exome sequencing on 9,381 individuals with at least one psychotic episode during their lifetime. SLCO1B1 SNVs were annotated with PHRED-scaled Combined Annotation-Dependent (CADD) scores and the Ensembl variant effect predictor (VEP). In vitro functionality studies were conducted for the SNVs with PHRED-scaled CADD score >10 and predicted to be missense. To estimate possible changes in transport activity caused by the variants, transport of 2’,7’-dichlorofluorescein (DCF) in OATP1B1 expressing HEK293 cells was measured. The amount of OATP1B1 in crude membrane fractions was quantified with a LC–MS/MS-based quantitative targeted absolute proteomics analysis.
Results Six rare missense variants of SLCO1B1 were identified in the study population located in transmembrane helix 3: c.317T>C (p.106I>T), intracellular loop 2: c.629G>T (p.210G>V), c.633A>G (p.211I>M), c.639T>A (p.213N>L), transmembrane helix 6: 820A>G (p.274I>V), and the C-terminal end: 2005A>C (p.669N>H). Of these variants, SLCO1B1 c.629G>T (p.210G>V) resulted in protein loss of function, abolishing the uptake of DCF and reducing membrane protein expression to 31% of reference OATP1B1.
Conclusions Of the six rare missense variants, SLCO1B1 c.629G>T (p.210G>V) causes loss-of-function of OATP1B1 transport and severely decreases membrane protein abundance. Carriers of SLCO1B1 c.629G>T may be susceptible to altered pharmacokinetics of OATP1B1 substrate drugs and may have increased likelihood of adverse drug effects such as statin-associated musculoskeletal symptoms.
Introduction
Statins are among the most common prescription drugs in the world and are used for the reduction of low-density lipoprotein cholesterol concentration and prevention of cardiovascular disease. The solute carrier organic anion transporter family member 1B1 (SLCO1B1) gene encodes the organic anion transporting polypeptide 1B1 (OATP1B1), a transmembrane protein that is involved in the transport of drugs such as statins and other compounds from the blood into the hepatocytes1,2.
Single nucleotide variants (SNVs) in SLCO1B1 are associated with impaired transporter function that can alter systemic exposure to statins. This can consequently predispose patients to adverse drug effects including statin-associated musculoskeletal symptoms (SAMS) and even rhabdomyolysis, impacting statin adherence and hindering the long-term effectiveness of statin therapy3,4,5,6,7. Cardiovascular mortality is one of the leading causes of excess mortality in patients with schizophrenia8. Antipsychotic medications, especially second-generation antipsychotic medications such as clozapine and olanzapine, can induce metabolic abnormalities including dyslipidemias9. Therefore, statins are commonly prescribed to patients with psychotic disorders.
The U.S. Food and Drug Administration (FDA) and European Medicines Agency (EMA) acknowledge the significance of OATP1B1 variants in medication safety by providing guidelines for studying the drug transporter’s effect on pharmacokinetics in drug development10,11.
Pharmacogenetic research has suffered from small sample sizes, and finding a patient group of sufficient size is often challenging when exploring rare genetic variants12. Rare variants of SLCO1B1 are mainly unreported although their effect on drug safety can be as consequential as the effect of common variants13,14. Interpretation of genetic variants from vast amounts of whole exome sequencing data remains a challenge and computational tools are needed to predict the functional impact of pharmacogenetic variants before the identified variants can be explored at a biological level15,16,17. Various algorithms that are needed to predict the functional impacts of coding variants are available using standard criteria and scoring metrics or machine learning approaches. Biological assessment of in silico evaluated variants is necessary to generate clinically actionable recommendations.
We studied the potential clinical relevance of rare SLCO1B1 SNVs occurring among 9,381 Finnish individuals with a psychotic disorder recruited as part of the SUPER-Finland study. The variants were selected for in vitro expression and functionality experiments based on a damaging in silico prediction by the PHRED-scaled Combined Annotation-Dependent (CADD) score algorithm18,19 and an Ensembl variant effect predictor (VEP)20 annotation of missense. Cellular uptake of the substrate 2,7-dichloro- fluorescein (DCF) in OATP1B1-overexpressing HEK293 cells was determined and the amount of OATP1B1 variant protein expressed in HEK293 cells was quantified with an LC–MS/MS based quantitative targeted absolute proteomics (QTAP) analysis. Our results provide functional annotations for SLCO1B1 c.317T>C (p.I106T), c.629G>T (p.G210V), c.633A>G (p.I211M), c.639T>A (p.N213K), c.820A>G (p.I274V) and c.2005A>C (p.N669H) variants.
Material and methods
SUPER-Finland study
The SUPER-Finland study recruited 10,474 participants aged >18 with a severe mental disorder from Finnish in- and outpatient psychiatric care, primary care, housing units and additionally with newspaper advertising between the years 2016-2018. Subjects with a diagnosis of a schizophrenia spectrum disorder (ICD-10 codes F20, F22–29), bipolar disorder (F30, F31) or major depressive disorder with psychotic features (F32.3 and F33.3) were included in the study. Exclusion criteria were inability to give informed consent and age under 18 years. We put special focus on ensuring wide coverage of known Finnish internal population subisolates21. The SUPER-Finland study protocol will be described in a separate cohort profile manuscript that is in preparation.
DNA was extracted from participants’ blood samples collected by venipuncture (2x Vacutainer EDTA K2 5/4 ml, BD). A saliva sample (DNA OG-500, Oragene) was collected for DNA extraction when venipuncture was not possible. The samples were frozen (−20 °C) within 60 min of sampling and sent to THL Biobank (Finnish Institute for Health and Welfare) within 3 months for long term storage (−185 °C). PerkinElmer Janus chemagic 360i Pro Workstation with the CMG-1074 kit was used to extract DNA from the EDTA-blood tubes. Extraction of DNA from saliva samples (after incubation +50 °C, o/n) was performed by Chemagen Chemagic MSM I robot with the CMG-1035–1 kit. The DNA samples were genotyped and sequenced at the Broad Institute of MIT and Harvard, Boston Cambridge, Massachusetts, USA.
Exome sequencing of SUPER-Finland study samples
To discover rare SLCO1B1 SNVs with possible changes in OATP1B1 protein function, we used exome data from 9,381 SUPER-Finland study participants. Exome sequencing was carried out with the Illumina HiSeq platform using 151 base pair paired-end reads at the Broad Institute of Harvard and MIT. The samples were enriched with the Illumina Nextera capture kit and sequenced until 80% of the target capture was covered at 20x. The Picard sequence processing pipeline was used to process BAM files (http://broadinstitute.github.io/picard/) and the data was mapped to the human genome reference build 38 (GRCh38) using BWA22. This procedure followed standard best practice alignment and read processing protocols as described earlier23,24. Variants were called using the Genome Analysis Toolkit (GATK25,26). Local realignment around indels and recalibration of base qualities in each sample BAM were performed with GATK version 3.4. Each sample was called with HaplotypeCaller to create gVCF files containing every position of the exome with likelihoods for variants alleles or the genomic reference allele. All variants were annotated using the Variant Quality Score Recalibration (VQSR) tool in GATK version 3.6 resulting in a VCF with germline SNVs and indels for all samples used in the analyses. The variant joint calling corresponded to the pipeline used to create the GnomAD database23.
In silico analysis of SLCO1B1 variants in SUPER-Finland study
The variants of chromosome 12 were converted to Hail27 matrix table file format and annotated with Combined Annotation-Dependent Depletion18,19 (CADD, version 1.4/1.6) scores and with the Variant Effect Predictor20 (VEP, version 95) tool through Hail (version 0.2, reference genome GRCh38). The variants were filtered with Hail to include only those having a call rate >0.99, a HWE p-value >1e-10, an allele count >1, a genotype quality >20 and a depth >10. The SLCO1B1 variants with a PHRED-scaled CADD score >10, predicted as missense variants by VEP and not included in the current CPIC guidelines for SLCO1B1 were selected for further in vitro expression and functional analyses.
Preparation of plasmids carrying SLCO1B1 variants
Plasmids carrying the SLCO1B1 variants were created as described by Kiander et al.14. The reference SLCO1B1 gene used was Genebank™ accession number AJ132573.1 and the mutagenesis primers are described in Supplementary Table 1. GATC Biotech’s (Constance, Germany) sequencing service confirmed the presence of the SNVs in the plasmids. Baculoviruses carrying the reference SLCO1B1 gene, the variant SLCO1B1 genes and the previously cloned gene for enhanced yellow fluorescent protein (eYFP) as a negative control were produced as described earlier by Tikkanen et al28.
Cell culture and protein expression
HEK293 human kidney cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) and high-glucose, GlutaMax culture medium supplemented with 10% fetal bovine serum (FBS) at 37 °C, 5% CO2. The cells (0.5 × 106) were seeded in each well of 48-well plates (ThermoFisher Scientific™ Nunc™ coated with poly-D-lysine in-house) 24 hours prior to transduction with the baculoviruses. To stimulate the expression of proteins, sodium butyrate was added with the viruses at a final concentration of 5 mM (as per in-house optimization).
Cellular uptake assays
The cellular uptake assay was performed 48 hours post-transduction on a heated (37 °C) orbital shaker plate. For a 3-minute preincubation, transport buffer (500 µl of HBSS with 4.17 mM NaHCO3 and 25 mM HEPES adjusted to pH 7.4 with NaOH) replaced the medium in the wells. After the buffer was removed, cellular uptake began when 125 µl of 1 µM 2,7-dichlorofluorescein (DCF) in the transport buffer was added into the wells. The uptake was stopped after 15 minutes by aspiration of the test solution. Three-time wash with 500 µl ice-cold transport buffer followed and the cells were lysed with 125 µl 0.1 M NaOH. Fluorescence measurement (excitation 500 nm, emission 528 nm, bandwidth 5 nm) with the multimode microplate reader Varioskan LUX (Thermo Fisher Scientific, Vantaa, Finland) of the cell lysates was performed to quantify the DCF. 10 µl of cell lysate was mixed with 300 µl Coomassie Plus reagent and used for total protein amount quantification with absorbance analysis (595 nm) on Varioskan LUX.
Crude membrane extraction
Baculoviruses carrying either reference or variant SLCO1B1 and sodium butyrate (5 mM final concentration) were added to the HEK293 cells after the cells were cultured for 24 hours in T175 flasks. The cells were collected and centrifuged (3000 g, 15 min) after 48 hours and broken down with Dounce tissue homogenizer, resuspended in Tris-sucrose (TS) buffer (10 mM Tris-HEPES, 250 mM sucrose, pH 7.4) and kept on ice. After a 30-minute centrifugation the supernatant was separated and centrifuged (21,000 g, 4 °C, 99 min) again resulting in a pellet containing the crude cell membrane. The protein sample was suspended in TS buffer and quantified as previously described.
LC–MS/MS based quantitative targeted absolute proteomics (QTAP) analysis
The LC–MS/MS-based QTAP approach was used to quantify the absolute protein expression of OATP1B1 in the crude membrane preparations. The method for the protein sample preparation and quantitation with targeted LC–MS is described earlier14,29,30. OATP1B1 and Na+/K+ATPase signature peptides were quantified from 50 µg of crude membrane fractions. After denaturation and break-down of the tertiary structure of the proteins, the crude membrane preparations were digested first with 1/100 LysC endopeptidase and after that with 1/100 TPCK-treated trypsin. Previously used isotope-labeled peptide mixture (3 fmol/µg protein) served as an internal standard14. Quantification was conducted with 6495 QQQ MS with a 1290 HPLC system and AdvanceBio peptide Map Column, 2.7 µm, 2.1 × 250 mm (Agilent Technologies, Santa Clara, CA, USA) as described previously14,30. The SNVs did not alter the amino acids in the analyzed peptide sequences. The peak area ratios of the analyte peptides and their respective internal standards were compared with the Skyline application (MacCoss Lab Software, Seattle, WA). The results of OATP1B1 expression are presented as relative to Na+/K+-ATPase expression level and normalized to the reference OATP1B1 protein. Absolute amount of OATP1B1 in proteomics samples is presented in Supplementary Table 2 and Supplementary Figure 1.
Data analysis
The uptake of the DCF was normalized to the total protein amount. Uptake into eYFP-expressing cells, representing passive influx, was subtracted from uptake into OATP1B1 expressing cells, yielding active OATP1B1 mediated transport. The transport activity in OATP1B1 variant cells was then normalized to the cells expressing wild-type OATP1B1. The statistical significance of the differences in activity and expression levels was determined with one-way analysis of variance (ANOVA) with the Dunnett’s post hoc test for multiple comparisons (GraphPad Prism 6.05, GraphPad Software, San Diego, CA, USA).
Ethics
The Coordinating Ethics Committee of the The Hospital District of Helsinki and Uusimaa (HUS) gave a favorable ethics statement (202/13/03/00/15) for the SUPER-Finland study. Prior to inclusion, written informed consent was obtained and archived from each participant. Individual-level data was pseudonymized.
Data and code availability
The computer code used in the analysis of this study is available from the corresponding author on reasonable request and the SUPER-Finland study data is available from THL Biobank when released from the original study.
Results
We evaluated the functionality of six SNVs of OATP1B1. The SNVs with their genomic positions, amino acid changes and locations in OATP1B1 are shown in Table 1.
Genomic positions, amino acid changes and locations of the SLCO1B1 SNVs.
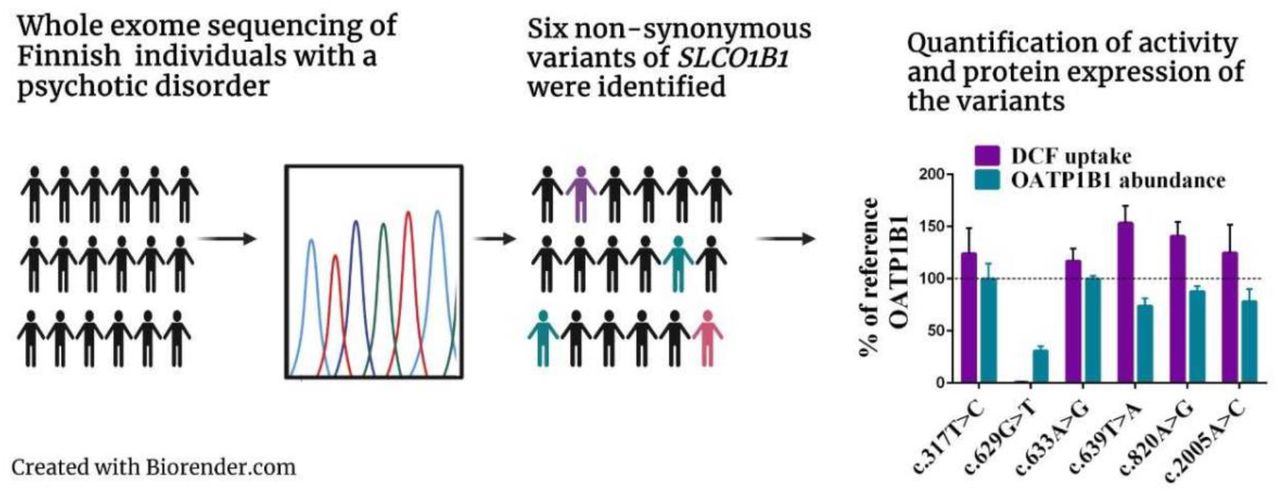
The variants were expressed in HEK293 cells and the effect of the SNVs on transport activity were evaluated with a cellular uptake study using DCF as a substrate. An LC-MS/MS-based QTAP approach quantified any changes in protein abundance of OATP1B1 in crude membrane fractions.
In silico predictions of SLCO1B1 variants in SUPER-Finland study
In silico predicted consequences of the six SLCO1B1 variants are presented in Table 2 and allele frequencies of the variants are shown in Table 3.
In silico prediction of consequences of the SLCO1B1 SNVs.
Prevalence of the SLCO1B1 SNVs.
SUPER-Finland subjects are heterozygous for the studied variants. The most frequent SNVs were c.639T>A, which was found in 16 subjects and c.317T>C, which was found in four subjects. The other four SNVs were found in only two subjects. Based on the in silico predictions by CADD, SIFT and PolyPhen, c.629G>T and c.2005A>C were most likely to have a damaging effect, and c.317T>C to be tolerated, while the predictions on the three other variants were more conflicting. All the variants are predicted to be missense variants by VEP. Two-dimensional prediction of the OATP1B1 transporter with the amino acid locations of the identified variants is presented in Figure 1.

Location of the amino acids affected by the identified SLCO1B1 SNVs in two-dimensional prediction of OATP1B1 transporter. Numbers 1-12 denote the putative transmembrane helices. The figure is based on the Uniprot entry Q9Y6L6 and generated with Protter31.
In vitro transport activity and membrane protein expression
We discovered that c.629G>T (p.G210V) abolishes the transport activity of OATP1B1 (Figure 2). Membrane protein abundance was also significantly decreased by approximately 70% compared to the OATP1B1 reference. The other variants did not alter transport activity or membrane protein expression to a statistically significant degree even though the activity was on average higher than the reference (Figure 2).

{kind=link}
{kind=link}
{kind=link}
OATP1B1 mediated transport of 1 µM DCF into HEK293 cells over 15 minutes: Results are calculated as mean of four experiments conducted in quadruplicates and represented % of reference OATP1B1 transport ±SEM (n=4). LC-MS/MS proteomics analysis of 50 µg HEK293 crude membrane preparations expressing variant OATP1B1: Abundance of OATP1B1 was quantified in four independent samples, normalized to Na+/K+-ATPase and reference OATP1B1 abundance (100%). The results are presented as mean ±SEM (n=4). **** = P<0.0001 (compared to the reference) ANOVA + Dunnett’s post hoc test.
Interestingly, though, c.639T>A (p.213N>K) seemed to enhance transport activity, which was on average 162% of reference OATP1B1 transport but failed to reach statistical significance. Protein expression of c.639T>A (p.213N>K), however, was not increased.
Discussion
We have provided novel functional annotations for six SLCO1B1 variants. Among these, c.629G>T (p.210G>V) was found to be a loss-of-function variant. The variant is in the intracellular loop of the OATP1B1 transporter, and according to GnomAD23 the variant has been detected only in Finnish and South Asian populations. We used computational algorithms to prioritize SLCO1B1 variants for in vitro validation. The PHRED-scaled CADD score for this variant was 23.4 and it was predicted to be deleterious by SIFT and probably damaging by PolyPhen. A variant with a similar CADD score of 23.0 (c.2005A>C, p.N669H) and classified as deleterious and possibly damaging by SIFT and PolyPhen, respectively, had normal function according to our in vitro studies. This underlines the difficulty of computational functional annotation of missense variants. Due to the enormous number of variants in whole exome sequencing studies, these studies need to rely on computational functional predictions, as not every potential loss-of-function variant throughout the genome can be tested in the laboratory. The definition of a computationally-predicted loss-of-function variant is a matter of sensitivity and specificity. Selecting a conservative definition would filter out potential disease-causing mutations, while too liberal a threshold creates noise in association signals. As CADD score is based on regional mutational constraint18,19, it might not be optimal in capturing pharmacogenetic variants as genes responsible for metabolism and transport of xenobiotic compounds might not be under as strong selection as genes related to, for example, brain development.
SLCO1B1 c.629G>T results in the replacement of a glycine with a valine in position 210, located at the end of the predicted intracellular loop 2 (Figure 1). Glycine has no side-chain and has a very high tendency to build turns in the secondary structure of polypeptide chains32. Valine, on the other hand, has a preference to form strands, and obstructs the formation of turns and bends in the secondary structure. Thus, it is understandable that this substitution can impair the proper folding and membrane localization of OATP1B1.
SLCO1B1 c.317T>C (p.106I>T), c.633A>G (p.211I>M) and c.820A>G (p.274I>V) are all in codons that originally code for isoleucine but the SNVs result in substitutions with amino acids with similar properties. Similar to isoleucine, valine and threonine have a preference for forming strands in the secondary structure of proteins. Additionally, methionine is neutral to strand formation and thus these variants most likely do not alter the secondary structure to a significant degree.
SLCO1B1 c.639T>A (p.213N>K) changes an amidic asparagine into a basic lysine, which in physiological pH has a positive charge. While the actual three-dimensional structure of the OATP proteins remain unknown, OATPs are predicted to form a positively charged pore33. Since many OATP1B1 substrates are anionic compounds, an additional positive charge near this putative pore in the intracellular loop where c.639T>A (p.N213K) is located might increase substrate affinity. According to GnomAD23 the c.639T>A variant has been identified only in Finnish population. A similar gain-of-charge occurs in SLCO1B1 c.1007C>G (p.P336R, located in TMH 7) and was shown in vitro to have substrate-dependent normal to enhanced OATP1B1 transport activity14,34. Likewise, SLCO1B1 c.2005A>C (p.669N>H) also gains a charge and increases activity slightly without increasing protein expression (Figure 2). All things considered, additional in vitro studies with multiple substrates and concentration-dependency assays are required to acquire supplementary mechanistic information on the effect of c.639T>A (p.213N>K) on OATP1B1 activity and its possible gain-of-function.
An increased-function phenotype has been reported for the SLCO1B1: *14 haplotype (that includes SNVs c.388A>G and 463C>A) increases OATP1B1 protein expression, decreases the exposure to atorvastatin and simvastatin acid, and enhances the LDL-lowering effect of fluvastatin35,36,37,38. Therefore, it is possible that the c.639T>A (p.213N>K) SNV characterized in this study might similarly increase hepatic intrinsic influx clearance of at least some substrate drugs in vivo, but this remains to be clarified. In the case of statins, where liver is the target organ, increased hepatic uptake and decreased systemic exposure are unlikely to have a negative impact. However, for substrates that are dependent on adequate systemic concentrations for their pharmacological action, such as methotrexate, a gain-of-function phenotype of OATP1B1 could reduce their efficacy and possibly require higher doses to reach the desired clinical outcome39.
Polypharmacy is common in patients treated with antipsychotic medication, leading to a high probability of adverse drug affects and drug-drug interactions40. While to date, according to The University of Washington Drug Interaction Database (DIDB)41, no central nervous system agents have been identified as OATP1B1 substrates, many other drugs can be prescribed to these patients to treat their somatic diseases. Among these are often statins to treat dyslipidemias which are associated with antipsychotic medications9. The association between statin-use and muscle symptoms has been thoroughly investigated. Variants in SLCO1B1 are associated with myopathy in particular when there is a documented increase in creatinine kinase concentration in blood3,42. A poor function phenotype of OATP1B1 can increase the probability for adverse effects during statin treatment thus reducing medication adherence.
The SLCO1B1*5 and SLCO1B1*15 haplotypes that contain the function-impairing c.521T>C SNV are associated with reduced hepatic clearance of substrate drugs, increased systemic exposure and increased risk of SAMS1,6,7. The current gene-based prescribing guideline from the Clinical Pharmacogenetics Implementation Consortium (CPIC) states that individuals with decreased and poor function phenotypes of OATP1B1 should limit statin doses and those with poor function phenotypes should avoid simvastatin altogether6. Based on our in vitro studies, c.629G>T (p.G210G>V) could be categorized as a poor function phenotype, since the in vitro activity and abundance is comparable to the SLCO1B1 c.521T>C genotype1,14. However, homozygous carriers of a rare variant like c.629G>T (p.G210G>V) are very uncommon. It is more likely to occur in a heterozygous state as observed in our study population (Table 3), or, like c.521T>C in SLCO1B1*15, in combination with c.388A>G, which has a frequency of approximately 26-77% depending on the population43. Additionally, c.521T>C allele is quite common in certain populations, for example the minor allele frequency in Finnish Europeans is 0.2123. Consequently, even as many as every fourth c.629G>T carrier is likely to carry c.521T>C resulting in a poor function phenotype. While the heterozygous genotype would limit the clinical consequences from a poor function phenotype to a decreased function phenotype, these individuals would still have an increased risk of SAMS during high-dose statin treatment and thus could benefit from genotype-guided dosing.
Conclusions
We have identified a novel SLCO1B1 loss-of-function mutation which could be accounted for in guidelines describing individualized lipid-lowering therapy. The variant c.629G>T (p.210G>V) abolishes the transport activity of OATP1B1 and may predispose patients to increased risk of SAMS during statin treatment. This emphasizes the need to include diverse populations in drug trials as adverse effects related to population-specific variants might otherwise be missed. The other five functionally-characterized SLCO1B1 variants (c.317T>C, c.633A>G, c.639T>A, c.820A>G and c.2005A>C) can be considered normal-function variants. Protein expression was not altered but DCF uptake was clearly enhanced, although failed to reach statistical significance, by the variant c.639T>A (p.213N>L). To study the possible gain-of-function effect on OATP1B1 activity of this variant, further in vitro studies with multiple substrates and concentration dependency assays are required.
Data Availability
The computer code used in the analysis of this study is available from the corresponding author on reasonable request and the SUPER-Finland study data is available from THL Biobank when released from the original study.
Author contribution
Conceptualization: M.N., A.V.A.; Data curation: K.H., W.K., K-S.V., L.U., J.L., SUPER-Finland researchers, A.V.A.; Formal analysis: K.H., W.K., H.K., M.N., A.V.A.; Funding acquisition: K.H., M.L., J.T., A.P., S.A., M.N., A.V.A.; Investigation: K.H., W.K., SUPER-Finland researchers, A.V.A.; Methodology: H.K., M.N., A.P., S.A., A.V.A.; Project administration : A.P., M.N., A.V.A.; Resources: H.K., M.N., J.T., S.A., A.P.; Software: K.H., L.U., J.L., A.V.A.; Supervision: H.K., M.L., J.T., M.N., A.V.A.; Validation: W.K., K-S.V., S.A.; Visualization: K.H., W.K.; Writing – original draft: K.H., W.K., H.K., M.N., A.V.A.; Writing – review & editing: K.H., W.K., H.K., M.L., L.U., K.-S.V., S.A., M.H., K.L., O.K., E.I., T.K., J.L., J.S., J.H., J.T., A.P., M.N., A.V.A.
Competing interests
M.L. is a board member of Genomi Solutions ltd., and Nursie Health ltd. and Springflux ltd., has received honoraria from Sunovion ltd., Orion Pharma ltd., Lundbeck, Otsuka Pharma, Recordati, Janssen and Janssen-Cilag. J.T. has participated in research projects funded by grants from Eli Lilly and Janssen-Cilag to his employing institution, has received honoraria from Eli Lilly, Evidera, Janssen-Cilag, Lundbeck, Otsuka, Mediuutiset, Sidera, and Sunovion, and is consultant to HLS Therapeutics, Orion, and WebMed Global. A.V.A is an employee and shareholder of Abomics, a company providing pharmacogenetic consultation services. The other authors declare no conflict of interest.
Funding
The SUPER-Finland study was funded by The Stanley Center for Psychiatric Research at the Broad Institute of MIT and Harvard, Boston, USA. K.H. has received funding from The Ministry of Social Affairs and Health Finland, through the developmental fund for Niuvanniemi Hospital, Kuopio, Finland, The Finnish Cultural Foundation, Helsinki, Finland, The Finnish Foundation for Psychiatric Research, Helsinki, Finland, The Social Insurance Institution of Finland, Helsinki, Finland, The Emil Aaltonen Foundation, Tampere, Finland, The Academy of Finland, Helsinki, Finland and The Yrjö Jahnsson Foundation (20207277), Helsinki, Finland. W.K. was funded by The Finnish Cultural Foundation, Helsinki, Finland and The Academy of Finland (332949), Helsinki, Finland. M.L. has received funding from The Finnish Medical Foundation, Helsinki, Finland and The Emil Aaltonen Foundation, Tampere, Finland. J.L. has received funding from The Finnish Medical Foundation, Helsinki, Finland and The Finnish Foundation for Psychiatric Research, Helsinki, Finland. M.H. has received funding from The Academy of Finland (310295), Helsinki, Finland. A.V.A. has received funding from The Orion Research Foundation, Espoo, Finland, The Juho Vainio Foundation, Helsinki, Finland and The Finnish Post Doc Pool. M.N. was funded by The European Research Council Consolidator Grant (grant agreement ID: 725249). The School of Pharmacy mass spectrometry laboratory in Kuopio is supported by Biocenter Kuopio and Biocenter Finland.
Acknowledgements
We thank the SUPER-Finland study participants. We would like to thank the collaborating SUPER-Finland study group: Mark Daly, Steve Hyman, Amanda Elliott, Benjamin Neale, Willehard Haaki, Teemu Männynsalo, Tuomas Jukuri, Kimmo Suokas, Jussi Niemi-Pynttäri, Asko Wegelius, Risto Kajanne, Aija Kyttälä, Noora Ristiluoma, Hannu Turunen, Auli Toivola, Annamari Tuulio-Henriksson, Tiina Paunio, Juha Veijola, Katriina Hakakari, Elina Hietala, Jaakko Keinänen, Martta Kerkelä, Tuomo Kiiskinen, Sari Lång-Tonteri, Nina Lindberg, Ville Mäkipelto, Atiqul Mazumder, Erik Cederlöf, Zuzanna Misiewicz, Julia Moghadampour, Arto Mustonen, Solja Niemelä, Olli Pietiläinen, Susanna Rask, Elmo Saarentaus, Anssi Solismaa, Andre Sourander, Marjo Taivalantti and Imre Västrik for the SUPER study data and support. We would like to thank Marju Monnonen, Natalija Trunkelj, Noora Sjöstedt and Laura Suominen for technical assistance as well as Dr. Yasuo Uchida for consultation on sample preparation of the proteomics samples. Dr. Mikko Gynther is acknowledged for the methodology in proteomics. We acknowledge the Drug Discovery and Chemical Biology Network for providing access to screening instrumentation and the Genomics Platform of the Broad Institute of MIT and Harvard for whole exome sequencing the SUPER-Finland study samples.
References



