
Introductory paragraph
The SARS-CoV-2 pandemic likely began by spillover from bats to humans1-3; today multiple animal species are known to be susceptible to infection4-8. White-tailed deer, Odocoileus virginianus are infected in the United States at substantial levels9-11, raising concerns about the formation of a new animal reservoir and potential of spill-back of new variants into humans12. Here we characterize SARS CoV-2 in deer from Pennsylvania (PA) sampled during fall and winter 2021. Of 93 nasal swab samples analyzed by RT-qPCR, 18 (19.3%) were positive for SARS-CoV-2. Seven whole-genome sequences were obtained, which were annotated as alpha and delta variants, the first reported observations of these lineages in deer, documenting multiple new jumps from humans to deer. The alpha lineage persisted in deer after its displacement by delta in humans, and deer-derived alpha variants diverged significantly from those in humans, consistent with a distinctive evolutionary trajectory in deer.
Main text
SARS-CoV-2 was detected by RT-qPCR in nasal swabs from 18 of 93 wild white-tailed deer sampled in Pennsylvania during fall and winter 2021 (19.3%; 95% CI: 12.6, 28.6) (described in Tables S1 and S2). Positive deer were identified in 10 of the 31 Pennsylvania counties surveyed from 10/2/2021 to 12/27/2021 (Figure 1). There was no difference in the proportion of positives between volunteer-collected samples and those collected by veterinary technicians (volunteer 11/66 and technician 7/27, p= 0.46). However, deer sampled as road-killed were significantly more likely to be positive than all other sample types (hunter harvested 11/66, road-killed 6/13 and other 1/14, p=0.002). The prevalence estimate was higher in female deer (35%; 95% CI: 22.13,50.5) than male deer (7.5%; 95% CI: 2.97,17.9). No information was available on possible symptoms or disease for the animals sampled.

Map of Pennsylvania (PA), showing sampling sites and locations of SARS-CoV-2 positive deer. The counties comprising PA are outlined. Counties with positive samples are shown in red; counties sampled but lacking positive samples are in blue; counties in grey were not sampled. The numbers of deer sampled and the number positive are shown for each county sampled. The deer samples sequenced were assigned to variants as indicated by the rectangles outside the map; variant type is color coded (teal for alpha/B1.1.7, purple/pink for delta/AY.#)
Viral whole-genome sequencing was attempted on the eight samples with the highest viral RNA levels (lowest cycle of threshold values in the RT-qPCR), and high-quality genome sequences were recovered from seven (Table S3). To confirm sample provenance, the non-viral sequence reads were analyzed for the deer samples and for human samples that were sequenced in parallel. All deer samples yielded reads mapping to the deer genome, and few or none mapping to the human genome. In contrast, reads from human samples overwhelmingly mapped to the human genome (Figure 2a), confirming the host species origin of our samples in deer.
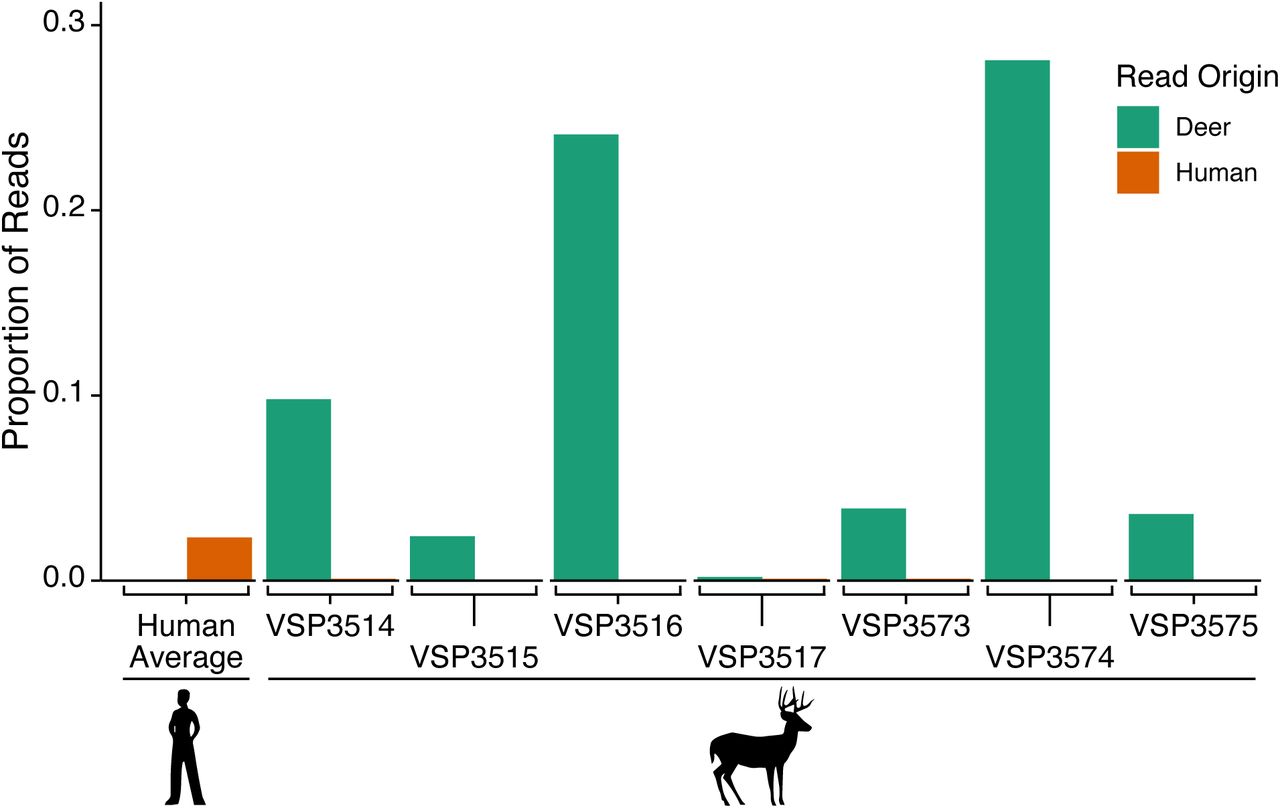
Checking sample tracking by analysis of nonviral sequences. Sequence reads from each SARS-CoV-2 sample were aligned to the white-tailed deer (GCF_002102435.1) and human (GCF_000001405.39) genomes, and nonviral reads enumerated. The numbers of deer and human reads are shown for each deer sample, denoted by the laboratory accession number VSP###. Results for 140 human samples, sequenced in the same sample batches, are shown to the far left. The proportion of reads aligning to the deer genome are shown in green, the average fraction aligning to the human genome is shown in orange.

{kind=link}
{kind=link}
{kind=link}
Analysis of SARS-CoV-2 whole genome sequences from white-tailed deer in PA and comparison to local human-derived sequences. A) Phylogeny of contemporaneous human alpha lineage sequences from PA, collected from 1/6/2021 to 11/16/2021, with deer sequences marked (1240 human and 2 deer sequences). B) Phylogeny of human delta lineage sequences from PA, collected from 10/14/2021 to 11/28/2021, with deer sequences marked (440 human and 5 deer sequences). C) Longitudinal comparison of deer variants and contemporary human variants. The bar plots show the progression of SARS-CoV-2 variants detected by genome sequencing in humans in eastern PA from 1/31/2021 to1/3/2021. The variants are color-coded according to the key to the right. The variants from white-tailed deer sequences are shown at the top of the figure, with the arrows showing the times of sampling. The deer isolates are color-coded on the deer icons as in the key.
Deer genomes annotated as alpha and delta variants, the first reported identification of these lineages in wild white-tailed deer. Previously the alpha variant was shown to be able to infect white-tailed deer that were experimentally inoculated11. Figure 2b shows a phylogenetic tree for alpha, and Figure 2c a phylogenetic tree for delta, in each case comparing Pennsylvania (PA) deer to contemporaneous human-derived sequences from across the same Mid-Atlantic region.
The two alpha sequences sampled from deer in adjoining counties in northeastern PA differed by 45 substitutions and were widely separated on the phylogenetic tree. A parsimonious explanation is thus that the two alpha lineages were introduced independently into deer and then diversified during transmission within deer. In addition, these deer-derived alpha genomes appear particularly divergent in contrast to the human alpha genomes sequenced previously in the same region. Only one out of 1240 human genomes were as divergent or more divergent from the inferred alpha common ancestor as were the deer sequence, giving a probability of randomly drawing two such divergent lineages as (1/1240)2=6.5×10−7.
For the delta variants (Figure 2C), two deer genomes were assigned to the same AY.103 clade and differed only by 7 substitutions, making the discrimination between a single or independent introductions for these sequences difficult. Two other deer SARS-CoV-2 genomes differed by over 21 substitutions from the AY.103 sequences and were assigned to the delta AY.88 lineage, which is an uncommon lineage for the PA area with only 0.14% of human-derived PA delta genomes being AY.88. The two AY.88 specimens from deer were identical. These likely represent a single introduction into deer given the scarcity of this lineage, the genetic similarity between samples, and the fact that the two isolates were from adjoining counties in northeastern PA and were sampled within a period of 2 days. The last delta sample was assigned to the AY.5 lineage. This genome clustered with the AY.88 specimens and differed by no more than 13 SNPs, making it unclear if it represents an independent introduction. Thus, a simple model would support identification here of at least four independent transmissions of SARS-CoV-2 from humans into deer, two of alpha and two of delta. It is also possible, though less likely, that SARS-CoV-2 entered deer fewer times and subsequently diverged, though this would require independent acquisition in deer of many mutations shared with contemporaneous human lineages.
The alpha and delta SARS-CoV-2 sequences from deer determined here were compared to deer SARS-CoV-2 sequences reported previously from other viral lineages to investigate possible deer-specific sequence polymorphisms. 108 deer genome sequences were downloaded from GISAID (Table S6), corresponding to samples acquired from 9/28/2020 to 2/25/2021 in Iowa and Ohio and dominated by early pandemic lineages including B.1.2 and B.1.3119,10. This revealed several substitutions that were highly enriched or invariant in deer isolates, but rare or absent in human isolates (Table S4). These include three silent mutations in ORF1ab, c7303t, c9430t, and c20259t. Mutation c7303t was found in 86% of these PA deer and 29% of previously published deer, whereas it was found in 0.04% of Mid-Atlantic human subjects and 0.09% of global genomes reported by NextStrain. Mutation c9430t was found in 43% of these PA deer and 56% of previously published deer, whereas it was found in 0.35% of Mid-Atlantic human subjects and 0.46% of global genomes reported by NextStrain. Mutation c20259t was found in 43% of these PA deer and 19% of previously published deer, whereas it was absent in Mid-Atlantic human subjects and present in 0.12% of global genomes reported by NextStrain (data from 1/26/2022). The enrichment of these mutations suggests possible functional interaction with deer-specific factors, which could influence RNA synthesis, RNA folding, or protein binding.
Samples from deer were compared longitudinally to samples from the mid-Atlantic region recovered from infected human subjects (Figure 2D). The alpha samples from deer were recovered in November of 2021, months after the alpha variant was displaced by delta in humans. In contrast, the delta isolates from deer were contemporaneous with the delta wave in humans. This suggests possible persistence of the alpha wave selectively in deer. Consistent with this, the deer alpha lineages appear relatively more diverged from human alpha isolates than do the deer delta lineages from human delta isolates (p=0.00056 for t-test comparing branch lengths of deer alpha versus delta isolates).
In summary, a survey of SARS-CoV-2 in 93 deer in Pennsylvania over the fall-winter of 2021 showed 19% of the deer sampled to be positive. Prior surveys carried out over the fall and winter of 2020 showed similarly high point prevalence of infection in Iowa (33.2%)9 and Ohio (35.8%)10. We report the first examples of the alpha and delta lineages in wild white-tail deer, likely derived from at least four independent spillovers from humans to deer. Given that there are estimated to be 30 deer square mile in PA, and over a million deer total, this suggests an enormous number of spillovers and infected deer in the state13,14. Mechanisms of infection and transmission are incompletely understood. Studies of experimental infections show efficient transmission between deer, potentially involving the common practice of individuals touching noses when in groups, but the mechanism of efficient transmission from humans to deer remains obscure. At present it is unclear to what degree SARS-CoV-2 is evolving in new directions in the deer host. Our findings of alpha persistence in deer after replacement of alpha by delta in humans, and the divergence seen between our deer and human alpha genomes, are all consistent with long-term persistence and spread of the alpha variant in deer.
Methods
Collection of samples from white-tailed deer
Samples were collected from hunter-harvested deer and sick or injured deer that were euthanized by state game wardens. Nasal swabs were taken within hours of death, placed in phosphate buffered saline (PBS) and stored in commercial refrigerators in field offices. Samples were shipped to the University of Pennsylvania within one week of collection and stored at -80°C until RNA extraction. Chi-squared tests were performed using R Statistical software (v4.0.5)15 (R Core Development Team 2021) to test for differences in proportions of positive deer by gender, age, source of deer (i.e., from volunteer compared to technician collectors as well as between hunter harvested, road-killed, or all other causes of death).
RNA extraction and detection of SARS-CoV-2 RNA by RT-qPCR
Nucleic acid was extracted with a QIAamp viral RNA mini kit (Qiagen) according to the manufacturer’s instructions. The presence of SARS-CoV-2 RNA was assessed by a RT-qPCR that specifically targets two different regions of the viral nucleocapsid gene as described previously.16
SARS-CoV-2 whole genome sequencing
Viral genomes were sequenced using the POLAR protocol17. The sample’s RNA (5μl) was mixed with Random Hexamers (0.5 μl of 50 μM, Thermo Fisher), dNTPs Mix (0.5μl of 10mM each, Thermo Fisher), and nuclease-free water (1 μl). This mixture was incubated (5 minutes at 65°C). Subsequently, reverse transcription was performed adding 6.5 μl from the previous reaction to SuperScript III Reverse Transcriptase (0.5 μl, Thermo Fisher), 5X First-Strand Buffer (2 μl, Thermo Fisher), DTT (0.5 μl of 0.1M, Thermo Fisher), and RNaseOut (0.5 μl, Thermo Fisher). This mixture was incubated (50 minutes at 42°C, then 10 minutes at 70°C). For cDNA amplification, the previous mixture (2.5 μl) was added to Q5 Hot Start DNA Polymerase (0.25 μl, NEB), 5X Q5 Reaction Buffer (5 μl, NEB), dNTPs mix (0.5 μl of 10mM each, NEB), and pooled Artic-ncov2019 v4 primers (reaction set 1 used 4.0 μl of pooled primer set 1, reaction set 2 used 3.98 μl of pooled primer set 2, IDT) and water (volume reaction brought to 25 μl). The samples were brought to 98°C for 30 seconds then cycled from 98°C for 15 seconds to 65°C for 5 minutes for 25 cycles, followed by 98°C for 15 seconds and 65°C for 5 minutes. The two reactions (one for each primer set) were pooled together. Tagmentation was completed with the Nextera XT Library Preparation Kit (Illumina). IDT for Illumina DNA/RNA UD Indexes were used for barcoding (Illumina). Quantification of DNA was performed using Quant-iT PicoGreen dsDNA quantitation assay kit (Invitrogen). The pooled libraries were quantified using the Qubit1X dsDNA HS Assay Kit (Invitrogen) and sequenced using the Illumina NextSeq platform.
Sequence Data Processing
Sequence data were processed as previously described18. BWA aligner tool (v0.7.17) was used to align subject sequences to the Wuhan reference sequence (NC_045512.2)19. Samtools package (v1.10) was used to filter alignments20. Variants were called using Pangolin lineage software (3.1.17 with the PangoLEARN 2021-12-06 release)21,22.
Host Sequence Analysis
The proportion of host sequences was inferred using a sampling of raw reads from all samples on any sequencing batch performed with deer specimen. 1,000 raw reads for each sample were blasted23-25 against a database constructed of a SARS-CoV-2 genome (NC_045512.2), human genome (GCF_000001405.39), feline genome (GCF_018350175.1), and white-tailed deer genome (GCF_002102435.1). Reads were tallied for each genome to which they were closely matched.
Mutation Analysis
108 deer-derived SARS-CoV-2 genomes were downloaded from GISAID and compared with the 7 deer sequenced in this publication and the human dataset. The previously published genomes were downloaded on 12/29/2021. Mutations were called in reference to the Wuhan strain (NC_045512.2).
Human subjects
Human sequences newly determined here were collected as follows. For most samples, the University of Pennsylvania Institutional Review Board (IRB) reviewed the research protocol and deemed the limited data elements extracted to be exempt from human subject research per 45 CFR 46.104, category 4 (IRB #848605). For hospitalized subjects, following informed consent (IRB protocol #823392), patients were sampled by collection of saliva, oropharyngeal and/or nasopharyngeal swabs, or endotracheal aspirates if intubated, as previously described 18. Further samples were collected from asymptomatic subjects detected in a screening program at the Perelman School of Medicine at the University of Pennsylvania and symptomatic subjects tested throughout the PennMedicine clinical network under IRB protocols #843565 and #848608. Human samples were sequenced as described for deer samples.
Phylogenetic Analysis
Deer and human sequences are available at GENBANK and GISAID (Table S3 and Table S7). NextClade was used to align sequences to the Wuhan reference26. IQ-TREE (v1.6.12) was used to infer a tree using maximum-likelihood methods using 1,000 bootstrap replicates27. Visualization of the inferred tree was performed using FigTree (v.1.4.4)28.
Data Availability
Sequence accession numbers for deer-derived SARS-CoV-2 genomes can be found in Table S3 (OM570187-OM570193) . Accession numbers for human SARS-CoV-2 genomes can be found in Table S7. Computer code is available at https://doi.org/10.5281/zenodo.4046252.
Data availability
Sequence accession numbers for deer-derived SARS-CoV-2 genomes can be found in Table S3 (OM570187-OM570193) . Accession numbers for human SARS-CoV-2 genomes can be found in Table S7. Computer code is available at https://doi.org/10.5281/zenodo.4046252.
Contributions
Conceived and designed the experiments. A. M., J. C. E., R. G. C., B. J. K., K. G. R., F. D. B., R. B. G., E. A.
Performed the experiments A. M., S. R., H. A., B. G., E. A.
Analyzed the data A. M, S. S.-M., J. E., F. D. B., B. G., E. A.
Contributed materials/analysis tools J. C. E., H. Z., S. S. G., C. C., K. S., R. G. C., B. J. K., K. G. R., F. D. B., R. B. G., E. A.
Wrote the paper A. M., S. S.-M., J. C. E., H. Z., S. S. G., C. C., K. S., R. G. C., B. J. K., K. G. R., F. D. B., R. B. G., E. A.
Supplementary Material
Supplementary Table 1. SARS-CoV-2 prevalence estimates stratified by sex, age, and sampling region.
Supplementary Table 2. Results of qPCR analysis.
Supplementary Table 3. SARS-CoV-2 whole genome sequences from white-tailed deer.
Supplementary Table 4. Single nucleotide polymorphisms in deer from this study.
Supplementary Table 5. Key materials and resources.
Supplementary Table 6. GISAID Acknowledgements.
Supplementary Table 7. Human Sequences Used.
Acknowledgements
We are grateful to hunters and wildlife personnel who provided specimens, and to Laurie Zimmerman for artwork and help with manuscript preparation. We acknowledge help from staff of the Philadelphia Department of Public Health. This work was supported in part by the Penn University Research Foundation. SG is supported by the Robert J. Kleberg, Jr. and Helen C. Kleberg Foundation. Funding was provided by a contract award from the Centers for Disease Control and Prevention (CDC BAA 200-2021-10986 and 75D30121C11102/000HCVL1-2021-55232), philanthropic donations to the Penn Center for Research on Coronaviruses and Other Emerging Pathogens, and in part by NIH grant R61/33-HL137063 and AI140442 -supplement for SARS-CoV-2. BJK is supported by NIH K23 AI 121485. Additional assistance was provided by the Penn Center for AIDS Research (P30-AI045008). HZ’s stipend to conduct this work was provided by a private donation by Tracy Holmes. This project has been funded in part with Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract No. 75N93021C00015.
Footnotes
Correcting typos in text
References



