
Abstract
Background Endometriosis is a common, complex disorder which is under-recognized and subject to prolong delays in diagnosis. It is accompanied by significant changes in the eutopic endometrial lining.
Methods We have undertaken the first single cell RNA-sequencing (scRNA-Seq) comparison of endometrial tissues in freshly collected menstrual effluent (ME) from 33 subjects, including confirmed endometriosis patients (cases) and controls as well as symptomatic subjects.
Results We identify a unique subcluster of proliferating uterine natural killer (uNK) cells in ME-tissues from controls that is almost absent from endometriosis cases, along with a striking reduction of total uNK cells in the ME of cases (p<10−16). In addition, IGFBP1+ decidualized subset of stromal cells are abundant in the shed endometrium of controls when compared to cases (p<10−16) confirming findings of compromised decidualization of cultured stromal cells from cases. By contrast, endometrial stromal cells from cases are enriched in cells expressing a pro-inflammatory phenotype. An enrichment of B cells in the cases (p=5.8 × 10−6) raises the possibility in some subjects of chronic endometritis, a disorder which predisposes to endometriosis.
Conclusions We propose that characterization of endometrial tissues in ME will provide an effective screening tool for identifying endometriosis in patients with chronic symptoms suggestive of this disorder. This constitutes a major advance, since delayed diagnosis for many years is a major clinical problem in the evaluation of these patients. Comprehensive analysis of ME is expected to lead to new diagnostic and therapeutic approaches to endometriosis and other associated reproductive disorders such as female infertility.
Brief summary The single cell analysis of menstrual effluent reveals differences in the endometrial tissues that may inform new diagnostic approaches to endometriosis.
Introduction
Endometriosis is a common and heterogeneous disorder that is characterized by the growth of endometrial-like tissues outside of the uterus, most commonly in the peritoneal cavity and associated with inflammation [1]. While the pathogenesis of endometriosis is not understood, retrograde menstruation of endometrial cells and tissues via the fallopian tubes is one accepted theory for the development of endometriosis lesions in the peritoneal cavity [2, 3]. However, retrograde menstruation occurs in nearly all women [4], yet endometriosis occurs in approximately one in ten females in their reproductive years [3]. Thus, other factors must contribute to the development of endometriosis. While there is a significant genetic component to endometriosis [5], very little is known about how these putative risk alleles function. On the other hand, the eutopic endometrium of patients with endometriosis is significantly different when compared to the endometrium of those without endometriosis, with inflammatory changes noted in the setting of endometriosis [6–9]. Given that endometrial tissue is transferred during retrograde menstruation and the endometrium of women with endometriosis is different from the endometrium of healthy controls [3–5], we have undertaken a detailed analysis of endometrial tissues and cells present in menstrual effluent (ME).
Most previous investigations of ME have involved the phenotypic analysis by immunofluorescence, flow cytometry, and/or in vitro culture of single cell suspensions collected using menstrual cups [10–14]. Our previous flow cytometry studies showed that uterine natural killer (uNK) cells were relatively depleted in ME from endometriosis cases vs. controls [11]. In addition, we demonstrated a defect in decidualization capacity of endometrial stromal cells grown from the ME of patients with endometriosis when compared to ME-stromal cells grown from healthy controls [11, 15]. While these earlier results potentially provided a basis for a screening test for endometriosis, these analyses relied on laborious and expensive cell culture and in vitro assays, making them impractical for clinical application.
Herein we investigated fresh ME as an unexplored and important biological specimen for the development of non-invasive diagnostics based on the direct analysis of endometrial tissue fragments. We show that ME contains large numbers of shed intact endometrial tissues. Using enzymatic digestion of ME and associated tissues followed by single cell RNA-sequencing (scRNA-Seq) analysis, we compared the major cellular differences and gene expression profiles found in ME collected from healthy controls and patients diagnosed with endometriosis, as well as patients with symptoms of endometriosis who are not yet diagnosed. In order to gain insight into the pathogenesis of endometriosis, we particularly focused on the phenotypes of stromal and uNK cells in ME through scRNA-Seq because these are abundant and have shown previously to be abnormal in eutopic endometrium of patients.
Results
Endometrial tissue fragments are present in fresh menstrual effluent
We carried out histological assessment of fresh menstrual effluent (ME)-associated tissues isolated from ME. Representative H&E sections of ME-derived tissue fragments from four subjects (1 control, 2 laparoscopically/histologically confirmed endometriosis subjects, and 1 symptomatic subject) show the presence of endometrial tissues with mucosal and glandular epithelium and areas of stroma. The endometrium had typical late secretory/menstrual morphology with expanded stroma containing scattered inflammatory cells, and secretory and inactive-type glands (Figure 1A-D, upper panels).Immunostaining of ME-derived tissue sections reveals an abundance of both stromal cells stained with antibodies to CD10, a clinically used marker of endometrial stroma [16, 17], and uterine NK cells (stained with antibodies to CD56 (NCAM), an archetypical marker of NK cells [Figure 1A-D, lower panels]).

Histological analysis of endometrial tissues isolated from the menstrual effluent (ME) from 4 separate subjects: A) control subject, B-C) two subjects with pathologically confirmed endometriosis, and D) subject chronic symptoms of endometriosis (not yet diagnosed). Upper panels for A-D: H&E staining is shown in two panels at two magnifications for each individual: A) 40X (left) and 200X (right); B) 100X and 200X; C) 100X an 200X; and D) 100X and 200X. Sections show typical late secretory/menstrual endometrium with expanded stroma containing scattered inflammatory cells and secretory and inactive type glands. Lower panels for A-D: immunostaining with anti-CD10 and anti-CD56 antibodies to detect stromal cells (left) and uterine NK (uNK) cells (right), respectively, at 100X. Scale bars are shown in each image.
Single cell RNA sequencing (scRNA-Seq) of digested freshly processed ME reveals the presence of a heterogenous mixture of immune and non-immune cells
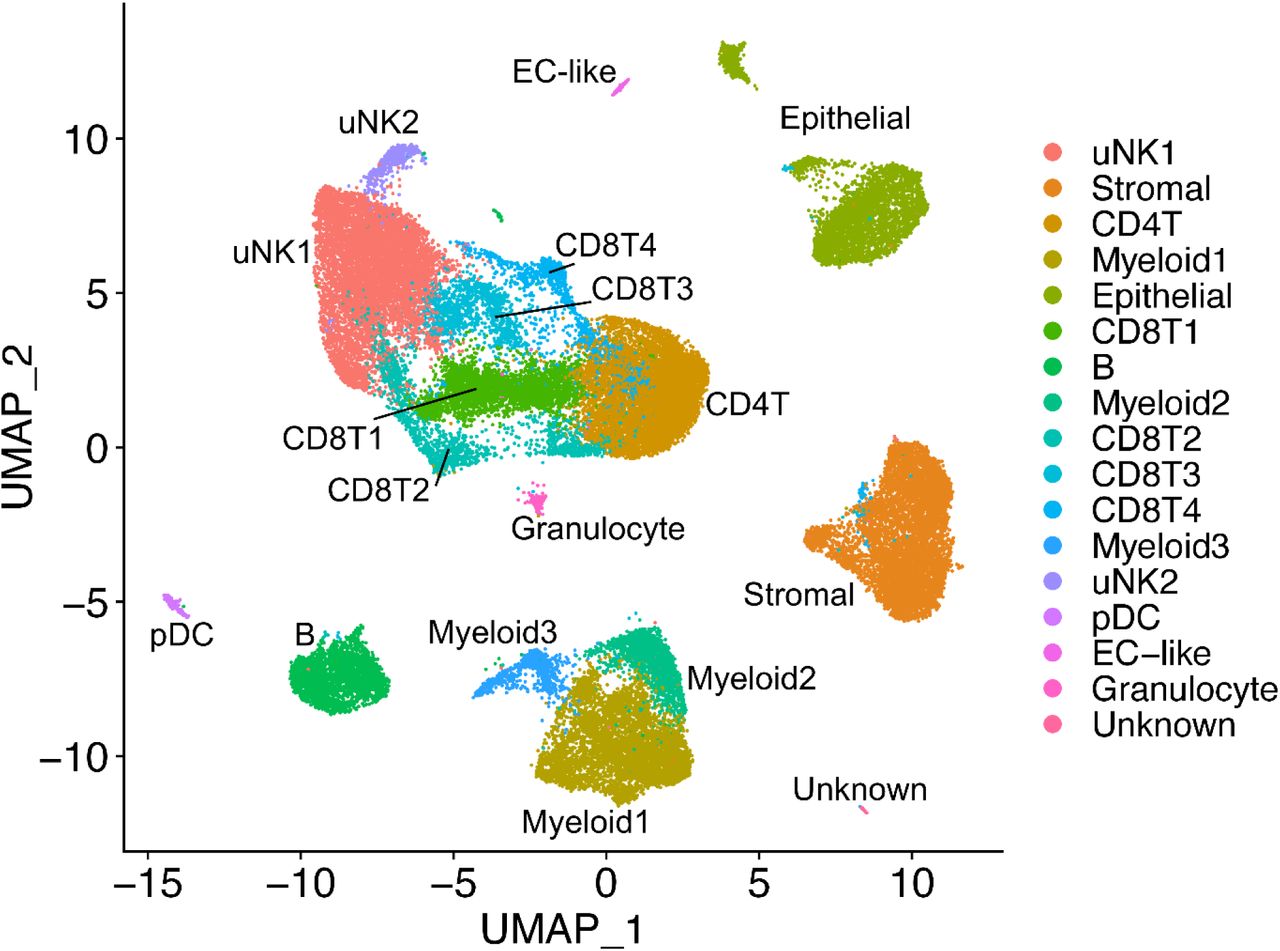
We have analyzed ME samples from 33 subjects, including age-matched healthy controls (N=9), endometriosis cases (N=11), and subjects with chronic symptoms of endometriosis but not yet diagnosed (N=13) (see Table 1). ME samples from either whole ME (unfractionated) or ME samples enriched for tissues (“ME-tissue”) were digested with collagenase I and DNase I, depleted of neutrophils, and processed for scRNA-Seq, as described in the methods. As shown in Figure 2 a graph-based clustering approach using Seurat distinguishes multiple cell clusters shown on the UMAP plot. There is striking diversity of the cell types defined by the cluster analysis. A major group of uterine NK cells is designated cluster uNK1, with a small associated cluster designated uNK2. Sets of clusters related to CD8+and CD4+ T cells are shown in the central portion of the plot. Endometrial stromal cells and epithelial cells are identified in major clusters in the right side of the UMAP plot, and distinct clusters of B cells and myeloid cells can also be delineated, along with a small cluster of plasmacytoid dendritic cells (pDC). It is important to note that these clusters are substantially similar whether unfractionated whole ME or tissue-enriched ME is processed for scRNA-Seq, except for epithelial cells, the yield of which is enhanced when tissue-enriched ME is utilized for sample processing (see Supplementary Figure 1).

UMAP plot for all 33 digested menstrual effluent (ME) samples (controls=9; endometriosis cases=11; symptomatic cases=13). Several well-delineated cell clusters include a large cluster of uterine NK cells (uNK1), as well as clearly separated stromal cells, epithelial cells, and B cells. Several clusters each of T cells and myeloid cells are also defined, as well as a small cluster of plasmacytoid dendritic cells (pDC). A small cluster of approximately 60 unknown cells is in the lower right corner.
Cell clusters from ME containing endometrial tissue differ between endometriosis cases and healthy controls; relative depletion of uterine NK cells and enrichment of B cells in endometriosis cases
We compared the frequency of the various cell clusters in the freshly processed ME obtained from the diagnosed endometriosis cases (N=11) compared with controls (N=9), as shown in Figure 3. By inspection of Figure 3, it is apparent that both clusters of uNK cells (uNK1 and uNK2) are markedly depleted in the cases vs. controls (average percentage of uNK approximately 8% in cases, 28% in controls), as well as an increase in the proportion of B cells in cases (~9%) vs. controls (~3%). The odds ratios and confidence intervals for these two cell enrichment patterns are shown in Figure 4, along with the patterns of enrichment of all the other major cell clusters. While there is some variation among many of the different cell clusters, a formal analysis shows the most striking differences are observed for uNK cells, which are enriched in controls (and depleted in cases; uNK1, P <10E-16; uNK2, P <10E-16), along with a relative enrichment in the proportion of B cells in the cases diagnosed with endometriosis (and relatively depleted in controls; P <10E-16). Note that the stromal cell cluster is not significantly different between cases and controls (P > 0.05).

The data taken from the UMAP plot in Figure 2 is separated into two groups: controls (n=9) and endometriosis cases (n=11). The most striking difference is the increased fractions of uterine NK cells (uNK1 and uNK2) in the endometrial tissues of controls as compared to cases. In contrast, B cells are significantly enriched in cases. A formal analysis of enrichment is given in Figure 4 and confirms the significant enrichment of uNK cells and B cells in controls and cases, respectively.

These data are taken from data shown in Figure 3. The Log2 odd ratios (OR) with cell subsets enriched in controls on the left and cell subsets enriched in cases on the right. It is appareent that uterine NK (uNK) cells, both uNK1 and uNK2, are significantly enriched in controls, while B cells show the greatest enrichment in cases. NOTE: Epithelial cells are excluded from this analysis because their enrichment was affected by the tissue preparation method used.
We also explored whether the various proportions of cell clusters of the ME preparations from the “symptomatic” but undiagnosed group of subjects (N=13) are different from ME preparations from controls. This is clearly the case, as shown in Supplementary Figure 2. Here, we show the relative enrichment of uNK cells is maintained in controls in comparison to the symptomatic group (uNK1, P <10E-16; uNK2, P = 0.0025), similar to that observed with ME from cases. B cells also show a significant relative enrichment in symptomatic as well as diagnosed cases, compared with controls (Supplementary Figure 2) (symptomatic vs. control, P = 5.8 ×10−6), similar to that observed with ME from cases. Perhaps not surprisingly, these significant differences in symptomatic cases vs. controls are less striking than the differences in endometriosis cases vs. controls, given the likely heterogeneity of the symptomatic group.
Decidualized stromal cell subclusters are reduced in endometriosis
Previous studies have reported reduced decidualization capacity in endometrial stromal cells grown from biopsies of patients with endometriosis [18]. We have also observed impaired decidualization using stromal cells grown directly from ME [11, 15]. Therefore, we examined whether this trend could be observed in fresh stromal cells analyzed by scRNA-Seq. The proportion of stromal cell numbers or percentages did not significantly differ between the control and endometriosis groups, as shown in Figure 3 and Figure 4. However, subclustering of the stromal cell cluster clearly identified 5 subclusters of interest within the stromal cell population (Figure 5A). We have designated these subclusters based on the dominant transcripts expressed in each of these subclusters, as shown in the violin plots in Figure 5B. The subclusters showing significant enrichment in either cases or controls are indicated by the Log2 (odds ratios, [OR]) below the UMAP plot (Figure 5C).

A) UMAP plot of the five stromal cell subclusters are shown. B) Violin plots showing the defining gene expression per subcluster for subclusters 1-5. C) Log2 (Odds Ratio) shows that subcluster 3 (IGFBP1+) is significantly enriched in controls (Log2 OR = −1.3, case vs. control). In contrast, subcluster 1 (IL11+) and subcluster 5 (MGP+) are enriched in controls. The IL11+ subcluster 1 and MGP+ subcluster 5 are significantly enriched in endometriosis. The top transcripts characterizing these three distinct stromal cell subclusters are summarized in Figure 6 and emphasize the enrichment of the decidualized stromal cells – subcluster 3 (IGFBP1+) – in controls.
It is striking that an apparently decidualized stromal cell subcluster (expressing IGFBP1 mRNA) is significantly enriched in controls compared with endometriosis cases (Figure 5A-B). In addition to IGFBP1, the top differentially expressed genes in this subcluster (compared to other stromal cell subclusters) include LEFTY2, DCN, LUM, MDK, C1QTNF6, APOE/D, DCN, and other progesterone sensitive and decidualization/fertility gene markers (see group 3 in Figure 6 and Supplementary Table 1). This suggests that a phenotype of “decidualization” can be measured directly in stromal cells derived from fresh ME and is associated with control vs. disease phenotype. A modest enrichment of a subcluster expressing IL11 was observed in cases, as indicated in Figure 5A-C. In addition to IL11, this subcluster is associated with transcripts for MMP3, MMP1, MMP9, SERPINB2, S100A6, and CXCL8, among other genes associated with inflammation, fibrosis and senescence, as well as endometriosis, as shown in Figure 6. A third subcluster, designated by high expression of the gene encoding matrix Gla protein (MGP), is also enriched in the stromal cells of cases (Figure 5A-C). This subset expresses numerous extracellular matrix genes that have been associated with presence of perivascular stromal cells, senescence, and cell adhesion/cell spreading, including FN1 (which encodes fibronectin-1), a known risk locus for endometriosis [19]. Figure 6 also shows the list of top genes expressed in this subset. Supplementary Figure 3 demonstrates that the IGFBP1+ and MGP+ subclusters map to stromal cells subsets defined in the decidua found in the first trimester of pregnancy by Vento-Tormo et al [20].

Distinct subclusters of decidualized stromal cells and pro-inflammatory stromal cells distinguish ME from controls and endometriosis cases. Upper panel: A summary of genes enriched in the stromal cell subclusters which are significantly enriched in cases (subclusters 1 [IL11+] and 5 [MGP+]) or controls (subcluster 3 [IGFBP1+]). Lower panel: Characteristic features of stromal cell subcluster gene markers. The decidualized stromal cell subcluster (IGFBP1+, subcluster 3) is prominently enriched in genes that are associated with decidualization and uterine receptivity and are progesterone responsive. In contrast, the non-decidualized stromal cell subsets that are enriched in cases (MGP+ [subcluster 5] and IL11+ [subcluster 1]) are variably enriched in estrogen responsive genes, and remarkably enriched in genes associated with inflammation, fibrosis, and cellular senescence. Note: MGP+ (subcluster 5) is also enriched in cell adhesion and cell spreading gene markers. See supplementary table 1 for references.
Finally, we examined the differences between cases and controls in the two uNK subclusters present in digested endometrial tissues in ME (uNK1 and uNK2, see Figure 2). We noted a distinct subcluster of uNK cells (uNK2) that is characterized by the expression of genes associated with cell proliferation such as MKI67 (which encodes Ki67) and TOP2A (which encodes topoisomerase 2A) (see Supplementary Figure 4 for a full uNK subcluster analysis). As discussed below, this cluster also mapped nearly exactly (97%) with a proliferative subset of uNK cells that has been defined by scRNA-Seq in decidua obtained during the first trimester of pregnancy [20]. This is consistent with the proliferation of uNK cells and overall accumulation of uNK cells in the course of decidualization in control subjects vs. cases, as shown in Figures 3 and 4.
Discussion
These studies show for the first time that the phenotype of eutopic endometrial tissue shed into the menstrual effluent is distinct in patients with endometriosis compared to control subjects. There are three major observations. First, the endometrial stromal cells show a relative deficiency of progesterone-sensitive gene markers associated with endometrial stromal cell decidualization in patients with endometriosis (e.g. IGFBP1, LEFTY2, LUM, DCN, etc). This is consistent with previous studies showing impaired decidualization of cultured endometrial stromal cells obtained from endometrial and ectopic endometriosis biopsies [18, 21], as well as from menstrual effluent [11, 15]. Secondly, there is a striking reduction in the proportion of uNK cells in the ME-derived endometrial tissue of patients with endometriosis compared with controls. This was suggested by our previous studies of free cells present in ME using protein-based flow cytometry methods [11], but it is clearly a major distinguishing feature of the eutopic endometrium of patients with endometriosis at the time of menstruation. Thirdly, our data suggest an enrichment of B cells in the eutopic endometrium of patients with endometriosis, a finding that is consistent with the hypothesis that chronic inflammation and/or chronic endometritis is a predisposing factor in the development of endometriosis [22]. Finally, our data support similar significant differences between controls and symptomatic cases who are not yet diagnosed.
A deficiency in the decidualization capacity of stromal cells cultured from biopsies of the eutopic endometrium has been reported previously [18], and is also found in ME-derived stromal cells collected at the time of menstruation [11, 15]. Our scRNA-Seq data clearly shows the reduction of the IGFBP1+-expressing decidualized stromal cell subclusters in endometriosis cases vs. controls (Figure 5C). The relationship of this finding to the pathogenesis of endometriosis is not established. One possibility is that a deficiency in this differentiation step leaves behind non-decidualized endometrial stromal cells that exhibit proinflammatory, pro-fibrotic, and/or senescent phenotypes. These ‘pathogenic’ cells may then initiate or promote lesions following retrograde transfer into the peritoneal cavity. The presence of the enrichment of an IL11-expressing stromal cell subcluster in the endometriosis ME samples that express many estrogen-responsive pro-inflammatory, pro-fibrotic and senescence gene markers in this study (Figure 6 and Supplementary Table 1) provides some support for this possibility, but this needs confirmation in larger datasets. The significant increase in the MGP+ stromal subcluster in endometriosis (Figure 5c) is also of potential interest. As shown in Figure 6, the MGP+ stromal cell subcluster expresses many genes that are associated with the extracellular matrix, including FN1 (encoding fibronectin-1) which has been associated with an increased risk for endometriosis in GWAS studies [23]. Interestingly, most of the top markers found in the IL11+ subcluster and the MGP+ subcluster are either associated with senescence or directly induce senescence (e.g., IL11 and SERPINB2 [Figure 6]). Inflammation and senescence are key features of endometriosis and reduced uterine receptivity and infertility [24–26].
Another possibility is that the overall environment of the eutopic endometrium predisposes to reduced stromal cell decidualization, independent of any direct role or effect on stromal cell subsets in the disease. A chronic inflammatory endometrial environment might lead to, or be associated with, other changes that put individuals at risk for endometriosis. For example, the presence of chronic endometritis has been reported to be a significant risk factor for endometriosis [22, 27]; chronic endometritis is also associated with reduced stromal cell decidualization [28]. Interestingly, the presence of B cells in endometrial tissue, particularly plasma cells, is a requirement for the clinical diagnosis of chronic endometritis [22]. We note the significant increase in B cells in shed endometrium of endometriosis patients (Figures 3 and 4) and symptomatic subjects (Supplementary Figure 2) when compared to controls. This may reflect an inflammatory state, as B cells play an important role in mediating or regulating inflammatory and autoimmune diseases [29]. The numbers of B cells available for detailed analysis have not allowed us to fully understand the phenotype of these cells; this is an area for future study.
We have clearly demonstrated that uNK cells are remarkably depleted in the ME-derived endometrial tissues of patients with endometriosis. This may reflect compromised decidualization in these subjects. uNK cells are a characteristic feature of decidualizing tissues [30] and are also prominent in the decidua of early pregnancy [20]. The presence of proliferating uNK cells is a characteristic of both tissues (Supplementary Figure 4). To our knowledge, this is the first report of proliferating uNK cells found in ME. Crosstalk between stromal cells and uNK cells is a feature that promotes decidualization and uterine receptivity/placental vascular remodeling [31]. uNK cells do not appear to play a major role in decidualization in uNK deficient IL15 knockout mice [32]. However, it remains unclear whether uNK cells or stromal cells are the primary driver of the reduction in the extent of decidualizing tissues in the setting of endometriosis. However, uNK do play a role in the maintenance of decidual integrity as reported by Ashkar et al [33]. Brighton and co-workers emphasized the important role of uNK cells in clearing senescent decidual cells in the cycling human endometrium and their clearance proposed to be important for optimal fertility [34]. A lack of uNK cells in the endometrium may contribute to increased numbers of senescent cells observed in the stromal subclusters among endometriosis subjects and may contribute to endometriosis-associated infertility. However, it is plausible that a lack of decidualizing endometrial stromal cells (with concomitant reduced production of IL-15 and uNK chemoattractants) reduces the infiltration and proliferation of uNK in decidualizing zones. However, we did no observed evidence of this in the late decidualized stromal cells in ME. Since uNK cells are reported to play a role in infertility [30, 35], and infertility is a common feature of endometriosis, further analysis of the uNK subset will clearly be of interest.
It is encouraging that many of our findings in patients with pathologically confirmed endometriosis are also present in a substantial proportion of subjects with chronic symptoms that are suggestive of endometriosis, even in the absence of a confirmed tissue diagnosis. The delay in diagnosis of endometriosis is widely recognized as a major barrier in the management of this disease, with delays of up to a decade in some subjects before the disease is recognized [36]. In our view, it is imperative to carry out a prospective evaluation of subjects with symptoms suggestive of endometriosis, along with matched controls without such symptoms, and to then confirm a tissue diagnosis at subsequent laparoscopy. Such a study design will be required to establish the positive and negative predictive value of menstrual tissue analysis in a real-world clinical setting where an endometriosis screening test might be applied.
What should such a screening test involve? Even without a complex scRNA-Seq analysis, gene expression patterns among stromal cells or uNK cells (or specific stromal cell and uNK subsets) may provide useful biomarkers. An initial analysis of stromal cell clusters suggests that there are potentially useful gene expression differences comparing cases and controls in a focused analysis of these cells, (Supplementary Figures 5 and 6), or indeed other cell types that remain to be investigated. On the other hand, if it can be adapted to a clinical diagnostic test, scRNA-Seq of these tissues is likely to be the most informative approach, perhaps having more global utility to establish complex and heterogenous disease subtypes, as well as predicting or following response to therapy. Additional phenotypes that can be uncovered using scRNA-Seq analysis on larger populations may yet yield additional biomarkers that can be incorporated into a more targeted multivariate biomarker analysis for diagnostic purposes.
In any case, the integration of our findings into a unified picture of the pathogenesis of endometriosis will require additional scRNA-Seq studies of larger heterogenous populations, at different stages of disease development and include deeper analysis of T cells, B cells, myeloid cells, and epithelial cells. Abnormalities of the eutopic endometrium at various times of biopsy are a widely recognized feature of endometriosis [18, 37], and indeed these changes in the eutopic endometrium can be observed in baboon models after the induction of experimental endometriosis [38]. This raises the possibility that retrograde menstruation of abnormal endometrial tissue may not be the causative pathway for the disease in some cases.
Nevertheless, abnormal eutopic endometrium is a characteristic disease associated phenotype [6–9, 18, 34], and this can provide diagnostic value, regardless of the underlying causative pathway. In addition, it is likely that characterization of eutopic endometrium in menstrual effluent using scRNA-Seq approaches may allow for the identification of clinically meaningful disease subsets and as a means for assessing patients’ responses to therapies, as well as uterine-associated fertility status. For example, many of the genes that exhibit changes in cell subclusters are associated with both estrogen and progesterone regulation (Figure 6), and these differences could be used to guide or assess responses to hormonal therapies and for assessing aspects of uterine receptivity/fertility.
On the other hand, if disease causation is due to retrograde menstruation of abnormal endometrial tissues, it provides an opportunity to explore new therapies using cells derived from menstrual endometrial tissues and cells. For example, we have previously shown that stromal cells grown from ME have persistent differences in decidualization capacity, even after weeks in culture [15]; similarly, treatment of ME-derived stromal cells with TNF and/or IL-1β significantly compromises their subsequent decidualization capacity for weeks [15]. These phenotypic changes can be induced in stromal cells in normal subjects by exposure to inflammatory cytokines. If decidualization defects are due to the endometrial inflammatory environment, targeted anti-inflammatory therapies might reduce this risk. In this context, it is important to remember that endometriosis has a large genetic component, approximately 50% heritability [39]. Thus, an individual’s response to a relatively common, and even intermittent, inflammatory environment may be a trigger for abnormal stromal cell differentiation in these individuals. An understanding of the gene-environment interactions underlying this process may suggest additional therapeutic possibilities.
These data represent a first attempt to globally characterize the cellular diversity of endometrium that is shed at the time of menstruation. Many cell types will require more detailed studies in larger datasets, particularly regarding diversity in T cells, B cells and myeloid cells, as well as epithelial cells. We propose that a comprehensive assessment of cellular phenotypes in the tissues present in ME will open a new window on both diagnosis as well as preventive treatment for patients at risk for endometriosis as well as other uterine and reproductive disorders.
Methods
Human subjects and menstrual effluent collections
Menstrual effluent (ME) was collected as previously described [11, 15]. Briefly, women of reproductive age (N=33, age 20-45 years, average age 33.6 years) who were not pregnant or breastfeeding, who were menstruating, and who were willing to provide ME samples were recruited and consented to the ROSE study (IRB#13-376A) (https://feinstein.northwell.edu/institutes-researchers/institute-molecular-medicine/robert-s-boas-center-for-genomics-and-human-genetics/rose-research-outsmarts-endometriosis). Women with histologically confirmed endometriosis (determined following laparoscopic surgery and documented in a pathology report) were enrolled as ‘endometriosis’ subjects (N=11). Women who reported chronic symptoms consistent with endometriosis (e.g., recurrent dysmenorrhea, persistent abdominal bloating, dyspareunia, dysuria, and/or dyschezia), but not yet diagnosed with endometriosis (or not) were enrolled as ‘symptomatic’ subjects (N=13). Control subjects who self-reported no gynecologic history suggestive of a diagnosis of endometriosis were enrolled as ‘controls’ (N=9). Endometriosis, symptomatic, and control subjects collected their ME using an ‘at home’ ME collection kit for 4-8 hours on the day of their heaviest menstrual flow (typically day 1 or 2 of the cycle) with a menstrual cup (provided by DIVA International), except for one subject who collected ME using a novel menstrual collection sponge (as previously described [15]). After collection, ME was shipped priority overnight at 4°C to the laboratory for processing. ME collected from menstrual cups was mixed 1:1 with DMEM for processing. For the saturated menstrual collection sponge, ME tissue was collected after rinsing the sponges with PBS to collect cells and tissue. Demographic and health data for controls, endometriosis subjects, and symptomatic subjects for each ME processing method (digested whole ME and digested ME-tissue) are shown in Table 1.
Immunostaining of ME-derived tissue fragments
ME-derived tissue fragments were collected by pouring ME over a 70μ filter, and transferred to the clinical pathology lab for fixation, paraffin embedding and H&E staining. CD10 was chosen for immunohistochemical analysis because it is a sensitive marker of eutopic endometrial stroma [17] and because adjunctive use of CD10 immunostaining with H&E staining enhances the histologic detection of endometriosis [16]. CD56 was chosen because uterine NK cells stain brightly with CD56. H&E slides and immunostained slides were examined microscopically and imaged by a pathologist.
Processing menstrual effluent for scRNA-Seq analyses
Whole (unfractionated) ME (2.5-10ml) was digested with Collagenase I (1mg/ml, Worthington Biochemical Corporation, Lakewood, NJ) and DNase I (0.25mg/ml, Worthington Biochemical Corporation) at 37°C for 10-30 min using the GentleMACS™ Tissue Dissociator (Miltenyi Biotec, Cambridge, MA). After digestion, the sample was sieved over a 70μ filter and washed with DMEM 10%FBS to neutralize digestion enzymes; the flow through was sieved over a 40μ filter and washed with DMEM 10%FBS. After collecting the single cells (from the flow through) following centrifugation (350xg for 5 min), Neutrophils were removed using the EasySep™ HLA Chimerism Whole Blood CD66b Positive Selection Kit (STEMCELL, Cambridge, MA), according to the manufacturer’s protocol. The neutrophil pellet was frozen at −80°C and used as a source of subject DNA for genotyping (see below). The resultant cells were depleted of red blood cells using the EasySep™ RBC Depletion Reagent (STEMCELL), according to the manufacturer’s protocol, and then washed and subjected to density gradient centrifugation using Ficoll-Paque PLUS (Sigma-Aldrich, St. Louis, MO) to collect mononuclear cells, according to manufacturer’s directions. To collect ME-tissue, whole ME (2.5-10ml) was sieved over a 70μ filter and washed with DMEM; the ME-tissues trapped on the filter was collected and digested with Collagenase I (1mg/ml, Worthington Biochemical Corporation, Lakewood, NJ) and DNase I (0.25mg/ml, Worthington Biochemical Corporation) at 37°C for 10 min and processed as described above for whole ME, except without a density gradient centrifugation step. The resultant whole ME cells were enumerated, and viability was assessed using ViaStain™ AOPI Staining Solution and the Nexcelom Cellometer (Lawrence, MA).
Preparations with >80% viability were processed for scRNA-Seq. Cells were immediately fixed in methanol for scRNA-Seq, as described by Chen for peripheral blood mononuclear cells [40]). Briefly, cells were washed and resuspended in a 200μl Ca++ and Mg++-free PBS, followed by dropwise addition of chilled 100% methanol (800μl, final 80% w/v). Fixed cells were stored at −20°C for 20min and then stored at −80°C until used for scRNA-Seq. Optimization studied demonstrated no difference in scRNA-Seq using fresh vs. methanol-fixed cells.
Processing of samples for single cell sequencing
Methanol-fixed cells were removed from −80°C and placed on ice for 5 min before centrifugation (1000xg for 5 min). Methanol-PBS supernatant was completely removed and cells were rehydrated in 0.04% BSA + 1mM DTT + 0.2 U/ul RNase Inhibitor in 3X SSC Buffer (Sigma). An aliquot of fixed cells was stained with Trypan Blue and visualized under the microscope. The cells were counted and pooled from different donors at equal ratios, filtered using 35μ strainer (Falcon), recounted and brought up to a final conc. of 2,000 cells/μl and proceeded immediately for GEM generation and barcoding on a 10X Chromium using Next GEM 3’ v3.1 reagents (10X Genomics). Libraries were constructed following 10X Genomics’ recommendations and quality was assessed on a High Sensitivity DNA chip on a BioAnalyzer 2100 (Agilent) before loading (1.8 pM) and sequencing on an Illumina Nextseq 500 using a High Output kit v2.5 (150 cycles). A total of 43,054 cells were analyzed in this study.
Single cell RNA-Sequencing and Analyses and Statistics
Samples were converted from raw bcl files to gene by cell matrices using CellRanger 6.0 aligned to 10x Genomics’ GRCh38-3.0.0 reference. Individuals were demultiplexed via Demuxlet [41] using genotypes taken from SNPs on the Illumina GSAv3 genotyping array, run on DNA prepared from neutrophils isolated from ME. The thresholds in Demuxlet were adjusted to the expected doublet rate and those marked as doublets were removed. Downstream analysis and visualization were done using Seurat 4.0[42]. Genes were filtered out if they were expressed in less than 3 cells while cells were filtered out if they had > 10% mitochondrial reads, 500 < nUMI < 5000 and > 6000 unique features. Only ME-tissue samples with >500 cells were kept. Gene expression normalization and cell clustering was done using the SCTransform pipeline [43] with % mitochondrial reads regressed out and person specific batch effects corrected using Harmony[44]. Identification of cell clusters was done using known markers genes with differential gene expression calculated using a Wilcoxon rank sum test. Enrichment of cell clusters of specific phenotypes was done using MASC (https://github.com/immunogenomics/masc) with covariates of batch, number UMI per cell, percent mitochondrial reads and phase accounted for. All datasets will be deposited in the National Center for Biotechnology Information/GEO prior to publication.
Study Approval
All procedures for the collection of samples from research subjects were performed with the approval of the institutional review board (IRB) of the Feinstein Institutes/Northwell Health. All participants signed informed consent priori to enrollment.
Data Availability
All data produced in the present study are available upon reasonable request to the authors
Supplementary Figures and Tables

Comparison of UMAP plots for ME samples prepared by tissue enrichment of menstrual effluent (“ME-Tissue”) and tissue digestion is applied to unfractionated ME (“whole ME”). Except for an increased yield of epithelial cells in ME samples enriched for tissue prior to digestion (ME-Tissue), there do not appear to be significant differences in overall cell composition between the two approaches to ME preparation.

The combined UMAP plot shown in Figure 1 is split into controls (n=9), cases (n=11), and subjects with convincing symptoms of endometriosis but without laparoscopic tissue diagnosis – the “symptomatic” group (n=13). Comparisons of uterine NK (uNK) cell and B cell frequencies show that the symptomatic group is most like cases, and different from controls.

We have compared the mapping of stromal subclusters reported by Vento-Tormo [20], based on the analysis of decidua in the first trimester, with the mapping of stromal cell subclusters we have described in menstrual effluent (ME). Note that our IGFBP1+ subcluster maps almost identically to the decidualizing stromal cell subset dS2 defined by Vento-Tormo. In addition, our MGP+ subset shows a substantial overlap with dP2 and dP1 of Vento Tormo, subsets which are attributed to the perivascular stromal cells in first trimester decidua.
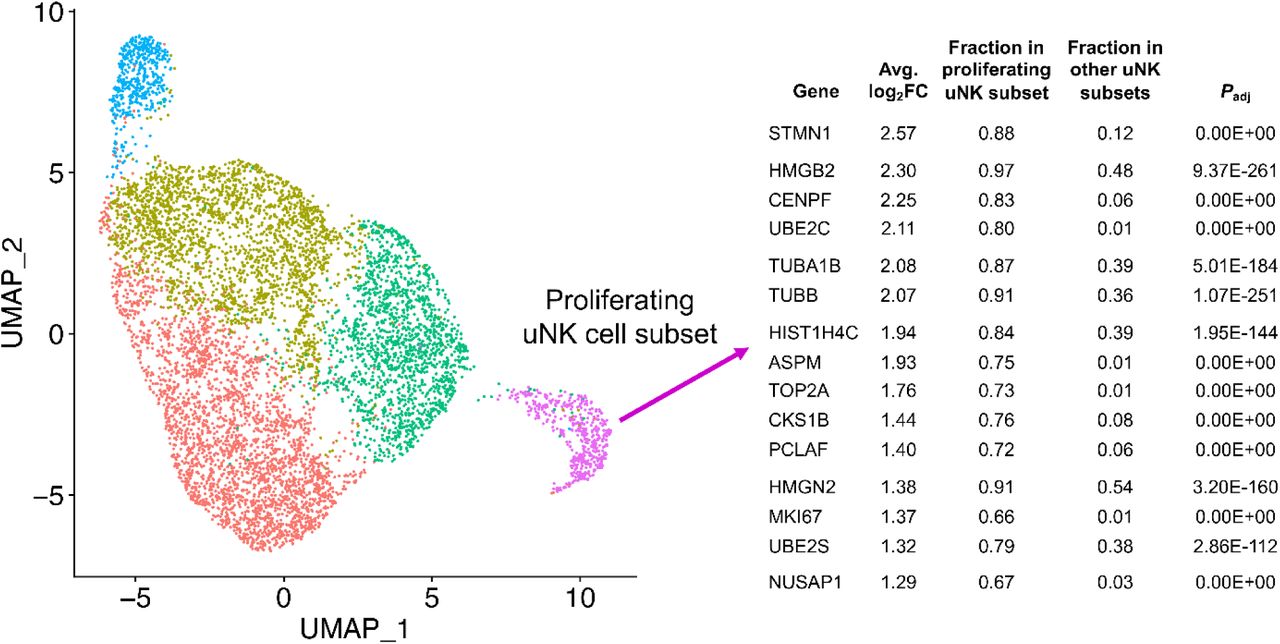
We have examined subclusters of uterine NK (uNK) cells in our dataset and identified a subcluster whose gene expression patterns reflect cell proliferation, with substantial enrichment of MKI67 and TOP2A. This subset is over 98% matched to a proliferative uNK cell subcluster defined in the decidua of first trimester pregnancy [20]. This subset corresponds to our subset uNK2 that is enriched in controls (Figure 3).
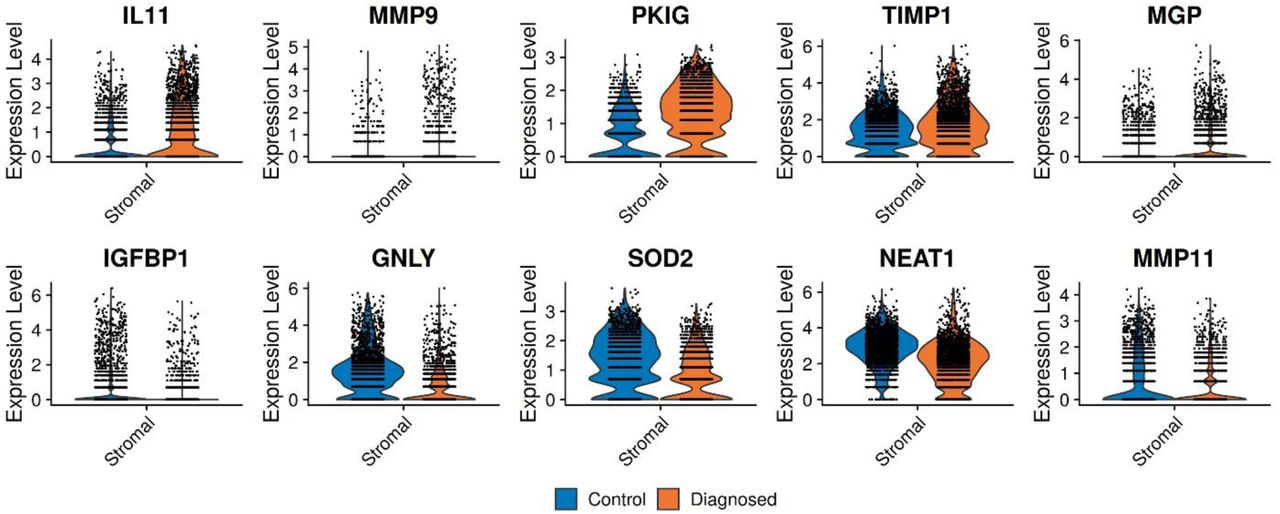
Violin plots of the top 10 genes that distinguish endometriosis cases and controls within the total stromal cell population in ME. The data suggest that IL11 and other transcripts may be useful in distinguishing stromal cells isolated from ME obtained from endometriosis case vs control subjects.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Violin plots of the top 10 genes that distinguish endometriosis cases and controls in an analysis of the uNK1 and uNK2 cell subsets as a whole. Note that IFITM2 and DNAJA1 expression are substantially higher in ME obtained from endometriosis cases and may provide a useful diagnostic target based on uNK cells that could be purified from tissues isolated from ME.
Acknowledgements
We are grateful to the Endometriosis Foundation of America and to Dr. Tamer Seckin for providing early support for this work, and the for the ongoing support from the Northwell Health Innovation Award. We also thank Anthony Liew, Cassie Pond and Maruf Chowdhury who provided valuable technical support for this project. We are especially grateful to the many extraordinary patients and volunteers without whose participation this project could not have been accomplished.
Footnotes
The authors have no conflicts of interest to declare.
This work was supported by the Northwell Health Innovations Award and the Endometriosis Foundation of America
References
References for Supplementary Table 1
- 1.
- 2.
- 3.
- 4.
- 5.
- 6.
- 7.
- 8.
- 9.
- 10.
- 11.
- 12.
- 13.
- 14.
- 15.
- 16.
- 17.
- 18.
- 19.
- 20.↵
- 21.
- 22.
- 23.
- 24.
- 25.
- 26.
- 27.
- 28.
- 29.
- 30.
- 31.
- 32.
- 33.
- 34.
- 35.
- 36.
- 37.
- 38.
- 39.
- 40.
- 41.
- 42.
- 43.
- 44.
- 45.
- 46.
- 47.
- 48.
- 49.
- 50.
- 51.
- 52.
- 53.
- 54.
- 55.
- 56.
- 57.
- 58.
- 59.
- 60.
- 61.
- 62.
- 63.
- 64.
- 65.
- 66.
- 67.
- 68.
- 69.
- 70.
- 71.
- 72.
- 73.
- 74.
- 75.
- 76.
- 77.
- 78.
- 79.
- 80.
- 81.
- 82.
- 83.
- 84.
- 85.
- 86.
- 87.
- 88.



