
Abstract
Background Acute respiratory distress syndrome (ARDS) with COVID-19 is aggravated by hyperinflammatory responses even after the peak of viral load has passed; however, its underlying mechanisms remain unclear. Alveolar epithelial injury is reported to be a very early event in ARDS with COVID-19. Herein, we assessed whether necrosis of alveolar epithelial cells and subsequent releases of damage associated molecular patterns (DAMPs) at an early disease stage aggravates ARDS with COVID-19
Methods We analyzed the levels of cytokeratin18-M65, an epithelial total cell death marker; CK18-M30, an epithelial apoptosis-specific marker; and HMGB-1, one of the DAMPs released from necrotic cells, in patients with COVID-19 with and without ARDS and healthy adults, in addition to the circulating alveolar epithelial and endothelial injury markers, namely sRAGE, angiopoietin-2, and surfactant protein-D. Molecular mechanisms of alveolar epithelial cell death and effects of neutralization on alveolar tissue injury were assessed using a mouse model mimicking COVID-19-induced ARDS.
Results COVID-19-induced ARDS was characterized by the elevation of sRAGE, an epithelial injury marker, at a very early disease stage. Although both serum levels of CK18-M65 and CK18-M30 were elevated in COVID-19-induced ARDS, the median CK18-M30/M65 ratio, an indicator of the fraction of apoptosis among total epithelial cell death, was 31.5% in serum from COVID-19 patients with ARDS, a value significantly lower than that of non-ARDS patients or healthy subjects. Moreover, the median M30/M65 ratio in bronchoalveolar lavage fluid (BALF) in COVID-19-induced ARDS was 27.8%, indicating that alveolar epithelial cell death is mainly caused by necrosis. Serum levels of HMGB-1 were also significantly elevated in ARDS versus non-ARDS patients. In a mouse model mimicking COVID-19-induced ARDS, the ratio of CK18-M30 to a total epithelial cell death marker in BALF was also lower than that in control subjects. Moreover, the alveolar epithelial cell necrosis involved two forms of programmed necrosis: necroptosis and pyroptosis. Finally, neutralization of HMGB-1 attenuated alveolar tissue injury in the mouse model.
Conclusions Necrosis, including necroptosis and pyroptosis, seems to be the primary form of alveolar epithelial cell death and subsequent release of DAMPs is a potential driver of COVID-19-induced ARDS.
Background
Infection with a novel strain of coronavirus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), causes coronavirus disease 2019 (COVID-19) pneumonia. In the most severe cases, the disease progresses to acute respiratory distress syndrome (ARDS), which is associated with severe alveolar tissue injury[1,2]. Interestingly, the disease severity is exacerbated by hyperinflammatory responses even after passing the peak of viral load[3,4]. However, the mechanisms that underlie disease aggravation in COVID-19-induced ARDS remain unclear. We and others previously reported that alveolar epithelial injury at a very early disease stage is a hallmark of COVID-19-induced ARDS[5,6], suggesting that alveolar epithelial injury may be a trigger of subsequent disease progression. Therefore, elucidating the detailed mechanisms by which alveolar epithelial injury occurs in COVID-19-induced ARDS may reveal a therapeutic target that prevents disease aggravation.
The alveolar epithelial injury in ARDS is characterized by cell death, which is divided into necrosis and apoptosis. Moreover, it has been recently reported that necrosis comprises not only accidental cell death but also several forms of programmed cell deaths[7,8]. Although previous studies have demonstrated that both alveolar epithelial necrosis and apoptosis are important for the pathogenesis of ARDS[9], a recent study in our lab demonstrated that necrosis is the primary form of alveolar epithelial cell death in lipopolysaccharide (LPS)-induced experimental ARDS[10]. In contrast to apoptosis, which does not elicit inflammation, necrosis causes the release of damage-associated molecular patterns (DAMPs) such as high mobility group box (HMGB)-1 from dead cells[11,12]. Therefore, it is possible that alveolar necrosis during early disease stages, and the subsequent release of DAMPs, may drive disease progression in COVID-19-associated ARDS[13,14].
Here, we assess whether alveolar epithelial cell necrosis and subsequent release of DAMPs aggravate COVID-19-associated ARDS. To determine alveolar epithelial cell death patterns in COVID-19 patients with or without ARDS, we analyzed serum levels of full-length (CK18-M65 antigen) and caspase-cleaved (CK18-M30 antigen) cytokeratin 18, which are epithelial total cell death and epithelial apoptosis markers respectively, in addition to the other several alveolar epithelial and endothelial injury markers. Moreover, we analyzed the levels of CK18-M65 and CK18-M30 in broncho alveolar lavage fluids (BALF) from COVID-19-induced ARDS. Next, we investigated the detailed mechanisms of alveolar epithelial cell death using the animal model mimicking COVID-19-induced ARDS[15,16]. Finally, we evaluated whether blockade of HMGB-1, one of DAMPs released from necrotic cells, can attenuate alveolar tissue injury in the animal model.
Some of the preliminary results of the study were published previously[5].
Methods
Clinical study design
In this single-center, retrospective, prospective observational study, we analyzed serum samples of adult patients with COVID-19 who were admitted to Yokohama City University Hospital from January 2020 to January 2021 and healthy controls matched as closely as possible for age and sex. Inclusion criteria for COVID-19 patients were, as follows: 1) a diagnosis of COVID-19 based on a positive real-time polymerase chain reaction test, 2) age ≥ 18 years, and 3) available residual serum samples. Additionally, we analyzed BALF samples from patients with COVID-19-induced ARDS who were admitted to the Yokohama City University Hospital from April 2021 to January 2022. ARDS was diagnosed based on the Berlin definition. The study protocol was reviewed and approved by the institutional review board of Yokohama City University Hospital (B200700100, B200200048). The requirement for informed consent was waived due to the observational nature of the study. Some preliminary data from retrospectively collected samples were previously published[5].
Clinical data collection
The following clinical data measured during the first 8 days of hospital admission were retrospectively collected from the medical charts of included patients: basal characteristics, vital signs, laboratory tests, and blood gas analysis findings.
Human serum sample analysis
Residual serum samples collected from patients with COVID-19 after daily laboratory tests were frozen for future use. Concentrations of human serum soluble receptors for advanced glycation end products (sRAGE) (DY1145, R&D systems, Minneapolis, MN), angiopoietin (ANG)-2 (DY623, R&D Systems), surfactant protein (SP)-D (DY1920, R&D Systems), cytokeratin (CK)18-M65 (M65 ELISA, #10040, VLVBio AB, Nacka, Sweden), CK18-M30 (M30-Apoptosense ELISA Kit, #10011, VLVBio), and HMGB-1 (#381-10531, Fuso, Osaka, Japan) were measured using commercially available enzyme-linked immunosorbent assay (ELISA) kits according to the manufacturer’s instructions. The ratio of CK18-M30/M65 was calculated, and when the value exceeded 100%, it was regarded as 100%.
Initial concentrations of these markers in ARDS and non-ARDS patients at admission (on the first or second hospital day) and healthy controls were compared. Further, temporal changes in levels of the markers were assessed in patients with ARDS throughout the 8-day period following hospital admission. In cases in which values were determined twice per day, mean values were used. In cases in which only a single value was available, the value was used.
Analysis of BALF samples from COVID-19-induced ARDS patients
BALF samples were obtained from six COVID-19-induced ARDS patients. A fiberoptic bronchoscope was wedged in a lateral or medial segmental bronchus of the right middle lobe, and lavage was performed using three aliquots of 50 mL of sterile isotonic sodium chloride solution. The collected BALF was centrifuged at 300 g for 5 min at 4 °C, and the supernatant was stored at −80 °C until analyses. The levels of CK18-M65 and CK18-M30 were quantified by ELISA, and the ratio of CK18-M30/M65 was calculated, as described above.
Animal experiments
All animal experimental protocols were approved by the Animal Research Committee of the Yokohama City University. Male specific-pathogen-free C57BL/6J mice aged 8–10 weeks that were purchased from Japan SLC (Shizuoka, Japan) were used for all animal experiments. Mice were housed under a 12-h light/dark cycle with food and water available ad libitum.
A mouse model mimicking COVID-19 was established based on previous reports.[15,16] Intratracheal administration of polyinosinic:polycytidylic acid (poly (I:C)) (P1530, Sigma-Aldrich, St. Louis, MO, USA) and the SARS-CoV-2 spike protein (Z03481, Lot B2103045, GenScript, Piscataway, NJ) was performed via the exposed trachea through a small incision at the front of the neck. During the procedure, mice were placed under general anesthesia using intraperitoneal ketamine and xylazine. Mice were euthanized 24 h after intratracheal instillation, and lung tissues and bronchoalveolar lavage fluid (BALF) samples were collected as previously described[10,17].
In a preliminary experiment, nine mice were randomly allocated into the following three groups (n = 3 per group): control, Poly (I:C), and Poly (I:C) combined with the SARS-CoV-2 spike protein. The poly (I:C) group received 250 μg intratracheal poly (I:C) dissolved in 100 μL phosphate buffered saline (PBS), and poly (I:C) combined with SARS-CoV-2 group received 50 μg SARS-CoV-2 spike protein with 250 μg poly (I:C) dissolved in 100 μL PBS. The control group received 100 μL PBS intratracheally.
Based on the results of preliminary experiments, mild and severe lung injuries mimicking COVID-19 were evaluated. Twelve animals were randomly allocated into the following three groups (n = 4 per group): control, mild COVID-19, and severe COVID-19. The severe COVID-19 group received 50 μg SARS-CoV-2 spike protein with 250 μg poly (I:C) dissolved in 100 μL PBS, whereas the mild COVID-19 group received 10 μg SARS-CoV-2 spike protein and 50 μg Poly (I:C) in 100 μL of PBS. The control group received 100 μL PBS intratracheally.
Finally, we evaluated effects of anti-HMGB-1 neutralizing antibodies on a severe COVID-19 animal model. Six animals were randomly allocated to anti-HMGB-1 antibody or isotype control groups (n = 3 per group). The severe COVID-19 animal model was established as described above. Then, 4 h after intratracheal instillation, 100 μg anti-HMGB-1 neutralizing (ARG66714, Arigo Biolaboratories, Hsinchu City, Taiwan) or isotype control antibodies dissolved in 100 μL PBS were intravenously administered via the tail vein under isoflurane anesthesia.
Analysis of the BALF of mouse models of COVID-19
Leukocytes from the BALF of mice were stained with Samson’s solution and counted. Protein concentrations in BALF were quantified using a bicinchoninic acid assay. Concentrations of sRAGE (DY1179, R&D systems), ANG-2 (MANG20, R&D systems), CK18-M30 (CSB-E14265M, CUSABIO, Houston, TX), total CK18 (CSB-E17158M, CUSABIO), and HMGB-1 (ARG81310, Arigo Biolaboratories) were measured using ELISA kits in accordance with the manufacturer’s instructions. Cytokines and chemokines were comprehensively analyzed using semiquantitative multiplex cytokine assay kits (ARY006, R&D systems) in accordance with the manufacturer’s instructions.
Western blotting
Total proteins from mouse lung tissues were extracted using trichloroacetic acid-acetone. Extracted proteins were solubilized. Thereafter, they were quantified using a bicinchoninic acid assay. Proteins were detected using primary antibodies against mixed lineage kinase domain-like (MLKL) (#28640, Cell Signaling Technology, Danvers, MA, dilution: 1:2000), pospho-MLKL (#37333, Cell Signaling Technology, 1:1000), gasdermin D (GSDMD) (ab219800, Abcam, Cambridge, UK, 1:3000), cleaved n-terminal GSDMD (A20197, ABclonal, Woburn, MA, 1:1000) and horseradish peroxidase-conjugated secondary goat anti-rabbit IgG antibodies (170-6515; Bio-Rad, Hercules, CA) as described previously[17]. Briefly, a certain amount of protein (MLKL: 5 μg, GSDMD: 2 μg, pMLKL: 30 μg, and cleaved GSDMD: 20 μg of proteins) was separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis, and transferred to polyvinylidene fluoride (PVDF) membranes. Equality of protein loading was confirmed by total protein staining (in case of protein amounts >10 μg) (Reversible Protein Stain Kit for PVDF Membranes, 24585, Thermo Fisher Scientific, Waltham, MA) or beta-actin (A5411, Sigma-Aldrich, 1:10000) staining (in case of protein amounts <10 μg). The density of each protein was determined using ImageJ software (National Institutes of Health, Bethesda, MD).
Histological analysis
Lung tissues of mice were fixed using 4% paraformaldehyde at 20 cm H2O pressure and embedded in paraffin for histopathological examination, as previously described[17]. Lung tissue sections were stained with hematoxylin and eosin for tissue injury evaluation. Lung tissue sections were stained with anti-pMLKL (ET1705-51, HUABIO, Woburn, MA, dilution: 1:100) and anti-GSDMD n-terminal (ER1901-37, HUABIO, 1:200) antibodies according to the manufacturer’s instructions.
Statistical analysis
Statistical analyses were performed using Prism 9 software (GraphPad, La Jolla, CA). Values of P < 0.05 were considered statistically significant. Data from clinical studies were presented as medians with interquartile ranges (IQRs), and analyzed as non-parametric data. Comparisons between basal characteristics and laboratory and physiological values among patients with and without ARDS were performed using Mann−Whitney or Fisher’s exact tests. Comparisons of the serum levels of tissue injury and cell death markers among patients with and without ARDS, and healthy controls were performed using Kruskal−Wallis analysis followed by Dunn’s multiple comparison test. Temporal changes in alveolar tissue injury markers were assessed using the Friedman test, and days in which alveolar tissue injury markers peaked were compared using Kruskal−Wallis analysis followed by Dunn’s multiple comparison test.
Data obtained from animal experiments were log-transformed and presented as means ± standard deviation. Data were analyzed using the Student’s t-test or one-way analysis of variance followed by Tukey’s multiple comparison test. Multiple cytokine and chemokine assays were analyzed using multiple t-tests via the false discovery rate approach comprised of the two-stage step-up method of Benjamini, Krieger, and Yekutieli[18]. The false discovery rate was set at 5%.
Results
Circulating alveolar tissue injury markers in COVID-19 ARDS
Forty-eight (18 non-ARDS and 30 ARDS) among a total of 84 patients hospitalized with COVID-19 during the study period and 18 healthy volunteers matched as closely as possible for age and sex, were included in the analyses of the circulating markers. Characteristics of patients with COVID-19 are presented in Table 1. Patients with ARDS had higher acute physiology and chronic health evaluation–II (APACHE-II) scores, white blood cell counts, C-reactive protein (CRP) levels, D-dimer levels, and lower ratios of partial pressure of arterial oxygen to fraction of inspired oxygen (P/F ratios) and lymphocyte counts than patients without ARDS. Eight patients with ARDS (26.7%) died. Among patients with ARDS, five developed acute kidney injury, with only a small increase in total bilirubin concentration was observed in several patients. Thus, organ dysfunction in most patients was primarily limited to the lungs.
The clinical characteristics in ARDS and non-ARDS patients with COVID-19.
We evaluated the circulating levels of three alveolar tissue injury markers: an alveolar epithelial injury marker (sRAGE) [19,20]and an endothelial injury marker (ANG-2)[21,22], along with an alveolar permeability indicator (SP-D)[23,24]. All the alveolar tissue injury markers levels after admission were significantly higher in patients with ARDS versus healthy controls (Fig. 1A–C). However, only serum sRAGE and SP-F levels of patients with and without ARDS significantly differed (Fig. 1A–C). In patients with ARDS, sRAGE levels were significantly elevated immediately after admission, and gradually decreased thereafter (Fig. 1D, G). On the other hand, ANG-2 and SP-F levels peaked later (Fig. 1E–G). Collectively, these results agree with prior work that demonstrated that severe alveolar epithelial cell injury at a very early disease stage is a hallmark of COVID-19-induced ARDS[5,6].
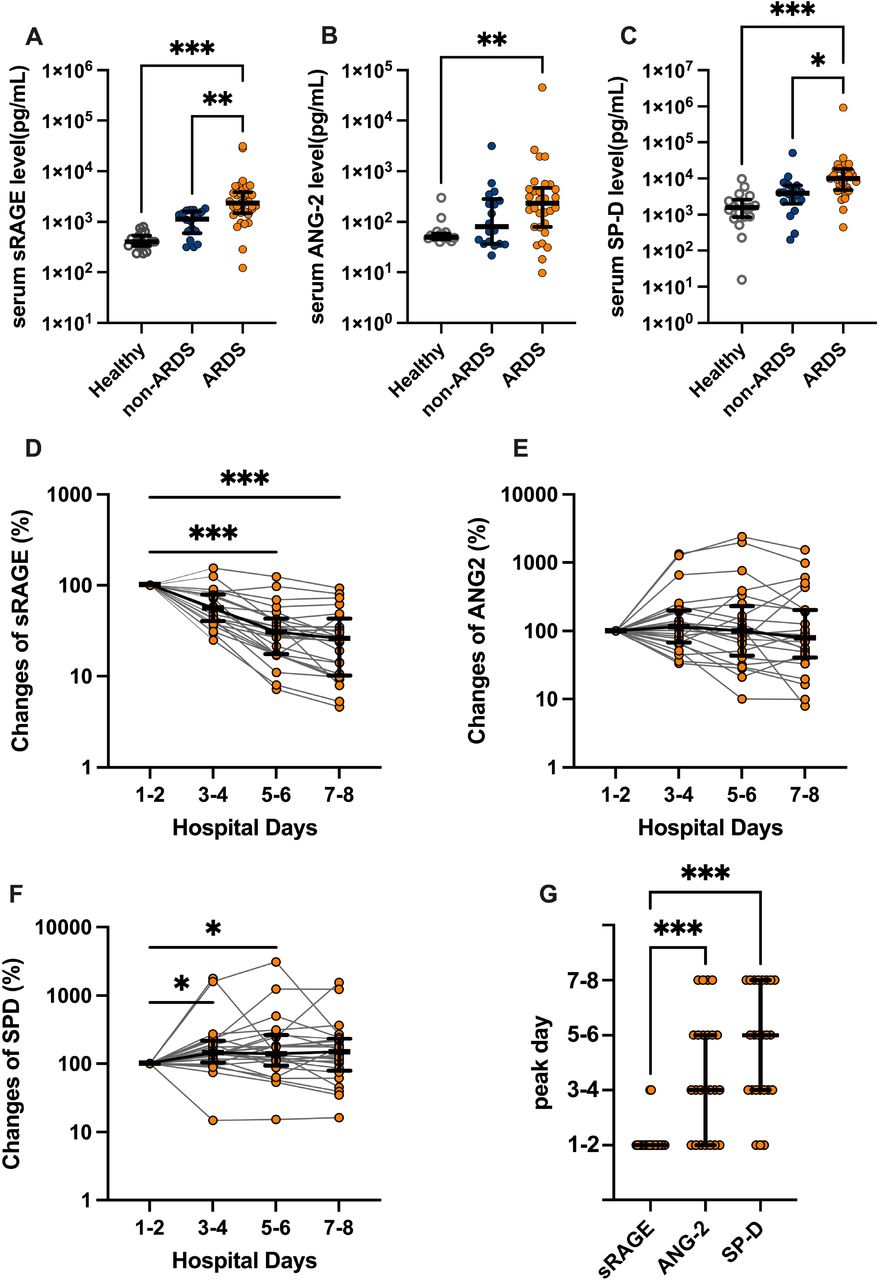
Analysis of serum levels of alveolar tissue injury markers using enzyme linked immunosorbent assays (ELISAs). (A) soluble receptors for advanced glycation end products (sRAGE), (B) angiopoietin (ANG)-2, and (C) surfactant protein (SP)-D levels in the serum of patients with COVID-19 with or without acute respiratory distress syndrome (ARDS) at admission (on the first or second hospital day), and healthy controls are shown. Bidaily temporal changes in (D) sRAGE, (E) ANG-2, and (F) SP-D in sera of COVID-19 patients with ARDS during first 8 days after hospital admission are shown. In cases in which multiple values every 2 days were available, mean values were used. When only a single value was available, the value was used. (G) Days in which concentrations of each alveolar tissue injury marker peaked in COVID-19 patients with ARDS are shown. Values are presented as medians with interquartile ranges. *p<0.05, **p<0.01, ***p<0.0001.
Epithelial necrosis markers and HMGB-1 are increased in COVID-19 ARDS
Next, levels of epithelial cell death markers were evaluated to elucidate the dominant form of alveolar epithelial cell death at early disease stage of COVID-19-induced ARDS. Serum levels of CK18-M65 and -M30 antigens were measured to distinguish alveolar necrosis from apoptosis. CK18 is exclusively expressed in epithelial cells, and is released upon cell death. The M65 antigen is an indicator of both epithelial cell necrosis and apoptosis. In contrast, the M30 antigen produced after caspase cleavage of CK18 is an indicator apoptotic epithelial cell death[25]. Although CK18 is expressed in all kinds of epithelial cells, it thought to be derived from alveolar epithelial cells in this cohort because the organ damage was limited almost exclusively to the lungs. Levels of both M65 and M30 at admission was positively correlated with disease severity in patients with COVID-19 (Fig. 2A, B). The M30/M65 ratio, which is an indicator of apoptosis, was significantly lower in ARDS patients [median: 31.5 %, IQR: 19.4−43.3] than healthy controls [median: 98.9 %, IQR: 83.1−100.0] or non-ARDS patients [median: 46.7 %, IQR: 36.6−80.5] (Fig. 2C). Moreover, we analyzed the CK18-M30/M65 ratio in BALF, which directly reflects the alveolar epithelial cell death, from six patients with COVID-19 ARDS (Supplementary Fig. 1). The characteristics of the included patients in the BALF analysis are shown in supplementary table 1. The M30/M65 ratio in the BALF was also low [27.8% IQR: 13.3−38.5] (Supplementary Fig. 1). Collectively, these results indicate that alveolar epithelial cell death in COVID-19 ARDS is mainly caused by necrosis.

Serum levels of markers of epithelial cell death and high mobility group box (HMGB)-1 in serum samples of COVID-19 patients with or without acute respiratory distress syndrome (ARDS) at admission (on the first or second hospital day) and healthy controls. Levels of (A) CK18-M30, an epithelial apoptosis marker, and (B) CK18-M65, an epithelial total cell death marker; (C) CK18-M30/M65 ratio, an indicator of the fraction of epithelial cells undergoing apoptosis versus all types of cell death; and (D) HMGB-1 levels in serum samples of COVID-19 patients with or without ARDS and healthy controls are shown. Values are presented as medians and interquartile ranges. *p<0.05, **p<0.01, ***p<0.0001.
Additionally, serum levels of HMGB-1, a kind of DAMPs released from necrotic cells, were significantly elevated in ARDS patients versus non-ARDS patients and healthy controls (Fig. 2D). The analysis of correlations among these biomarkers demonstrated that HMGB-1 levels were most strongly correlated with M65 levels, suggesting that epithelial cell death contributes to HMGB-1 release to circulation (Supplementary Fig. 2).
Intratracheal instillation of SARS-CoV-2 spike proteins combined with poly (I:C) to mice induces lung injury mimicking COVID-19-induced ARDS
It has been reported that innate immune responses to components of SARS-CoV-2 are principal drivers of inflammation and alveolar tissue injury in COVID-19[26–28]. To elucidate mechanisms of alveolar epithelial cell death in COVID-19-induced ARDS, we established animal models of severe and mild COVID-19 by intratracheal instillation with the SARS-CoV-2 spike protein and poly (I:C), a synthetic analog of double-stranded RNA based on previous reports [15,16] and our preliminary experiments (Supplementary Fig. 3). In the COVID-19 animal model, leukocytes infiltration (Fig. 3A), increased levels of protein, sRAGE, and ANG-2 in BALF (Fig. 3A–D), and lung tissue injury (Fig. 3E) were observed. Further, levels of several chemokines and cytokines previously reported to be elevated in COVID-19 patients[29,30] were significantly increased in the BALF of animal models of severe COVID-19 versus controls (Fig. 3F).

Use of a mouse model of mild and severe COVID-19. (A) White blood cell count, (B) total protein, (C) soluble receptors for advanced glycation end products (sRAGE), and (D) angiopoietin (ANG)-2 levels in bronchoalveolar lavage fluid (BALF) of mouse models of COVID-19 and controls are shown. (E) Representative lung tissue images of sections stained with hematoxylin and eosin are shown. Values are presented as means ± standard error. *p<0.05, **p<0.01, ***p<0.0001. (F) A heatmap constructed from the comprehensive analysis of cytokine levels in the BALF of mouse models of severe COVID-19 versus control mice. †q-value<0.05.
Necrosis, including necroptosis and pyroptosis, is a primary form of alveolar epithelial cell death in mouse models of severe COVID-19
Levels of Both CK18-M30 and total CK18, which is equivalent to CK18-M65, were increased in the BALF of COVID-19 models versus controls (Fig. 4A, B), indicating that both necrosis and apoptosis are involved in alveolar epithelial cell death. CK18-M30/total CK18 ratio, an indicator of apoptosis fraction relative to total epithelial cell death, decreased as lung injury increased in severity, as was observed in COVID-19 patients (Fig. 4C). Additionally, HMGB-1 levels were significantly elevated in severe COVID-19 animal model versus the other two groups (Fig.4D). Taken together, these results demonstrated that animal models of severe COVID-19 exhibit the same pattern of alveolar epithelial cell death as COVID-19 patients with ARDS.

Mechanisms of alveolar epithelial cell death in a COVID-19 mouse model. Levels of (A) CK18-M30 and (B) total CK18 in bronchoalveolar lavage fluid (BALF) from a COVID-19 mouse model are shown. (C) The ratio of CK18-M30/total CK18, an indicator of the fraction of apoptosis versus total epithelial cell death, is shown. (D) HMGB-1 levels in BALF from a COVID-19 mouse model. (E) Images and (F) densitometry of mixed lineage kinase domain-like (MLKL), p-MLKL, gasdermin D (GSDMD), and cleaved GSDMD immunoblots of the protein extracted from the lung of a COVID-19 mouse model are shown. (G) Representative images of the immunohistochemical analysis of p-MLKL and GSDMD in lung sections of mice are shown. Values are presented as means ± standard error. *p<0.05, **p<0.01, ***p<0.0001.
Next, we evaluated whether some forms of programmed necrosis are involved in alveolar epithelial cell death animal models of COVID-19. Necroptosis and pyroptosis are two types of programmed necrosis that are induced by inflammation[31]. In lung tissues of animal models of severe COVID-19, levels of phospho-MLKL and cleaved GSDMD, executioners of necroptosis[32,33] and pyroptosis[34], respectively, were significantly elevated versus the other two groups (Fig. 4E, F, Supplementary Fig. 4). Moreover, immunohistochemical analysis demonstrated that both phospho-MLKL and GSDMD are localized within alveolar walls (Fig. 4G). Collectively, these results indicate that necroptosis and pyroptosis contribute to alveolar epithelial cell death in COVID-19-induced ARDS.
Anti-HMGB-1 antibody treatment attenuates alveolar tissue injury in animal models of severe COVID-19
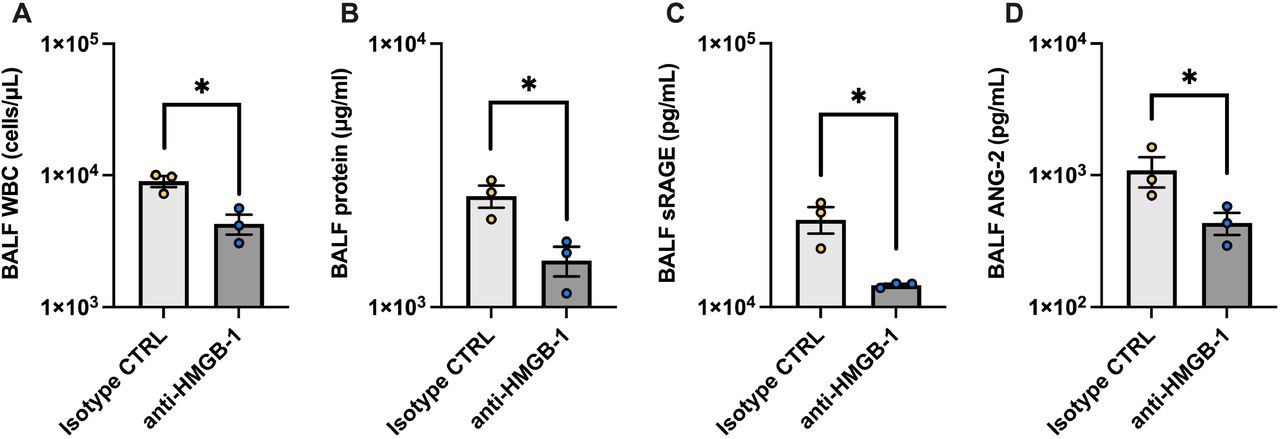
Necrosis of alveolar epithelial cells seems to occur in very early stages of the pathogenesis of COVID-19-induced ARDS, and it is difficult to prevent alveolar epithelial necrosis prior to hospital admission. Therefore, we evaluated whether inhibition of one of DAMPs, HMGB-1, attenuated alveolar tissue injury in a severe COVID-19 animal model. Treatment with the anti-HMGB-1 neutralizing antibody 4 h after intratracheal instillation of poly (I:C) and the SARS-CoV-2 spike protein significantly decreased BALF levels of leukocyte infiltration, total protein, sRAGE, and ANG-2 (Fig. 5 A–D). These results suggest that DAMPs such as HMGB-1 are promising therapeutic targets that may be used to prevent the aggravation of COVID-19-induced ARDS after hospital admission.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
An analysis of effects of high mobility group box (HMGB)-1 neutralization on alveolar tissue injury in a mouse model of severe COVID-19. (A) Total protein, (B) white blood cell count, (C) soluble receptors for advanced glycation end products (sRAGE), and (D) angiopoietin (ANG)-2 levels in BALF from the mouse model of severe COVID-19 treated with an anti-HMGB-1 neutralizing antibody or an isotype control antibody are shown. Values are presented as means ± standard error. *p<0.05, **p<0.01, ***p<0.0001.
Discussion
In the present study, we demonstrated that necrosis is the main form of alveolar epithelial cell death in COVID-19-induced ARDS. Moreover, two forms of programmed necrosis, necroptosis and pyroptosis, were involved in the alveolar epithelial necrosis in mouse models of severe COVID-19. Animal experiments also suggested that DAMPs released from necrotic alveolar epithelial cells such as HMGB-1 are potential drivers of disease exacerbation in COVID-19-induced ARDS.
Alveolar tissue injury in severe COVID-19 is aggravated after passing the peak viral load[3,4]. Therefore, hyperinflammatory responses only against SARS-CoV-2 per se cannot fully explain mechanisms that underly disease progression. Recently, we published findings suggesting that alveolar epithelial injury at a very early disease stage may trigger subsequent COVID-19 progression[5]. The present study indicates that initial alveolar epithelial necrosis, and the subsequent release of DAMPs may exacerbate alveolar tissue injury, which can progress even after viral loads have peaked.
Previously, it was shown that both necrosis and apoptosis are involved in alveolar epithelial cell injury in ARDS[9]. Because apoptosis can be easily assessed by TUNEL staining or caspase detection, the contribution of alveolar epithelial apoptosis in ARDS has been extensively studied[35]. However, we recently demonstrated that necrosis is the dominant form of alveolar epithelial cell death in LPS-induced ARDS by quantification of CK18-M30 and total CK18, which is equivalent to CK18-M65, in addition to cell labeling techniques[10]. In particular, quantification of CK18-M30 and M65 levels using commercially available ELISA kit can be applied for evaluation of epithelial apoptosis and necrosis in clinical setting. In fact, patterns of epithelial cell death such as sepsis[36,37] and graft rejection after lung transplantation[38] have been analyzed previously. This is the first study to suggest that necrosis is the primary form of alveolar epithelial cell death in human ARDS. Further studies will be needed to identify alveolar patterns of epithelial cell death in ARDS that is induced by disease etiologies other than COVID-19.
Necrosis was previously thought to cause accidental cell death. However, it has been demonstrated that some forms of necrosis, which are called as programmed necrosis, are regulated via molecular pathways[7]. Several animal studies have demonstrated that programmed necrosis is involved in alveolar epithelial cell death in ARDS[39–42]. Moreover, studies have suggested that SARS-CoV-2 activates intracellular necroptosis and pyroptosis pathways[31,43,44], and it has been reported that the circulating level of receptor-interacting protein kinase 3, a kinase required for necroptosis, is elevated in critically ill patients with COVID-19[45]. In line with findings of these studies, our animal experiments suggest that necroptosis and pyroptosis are involved in alveolar epithelial cell death in COVID-19 ARDS.
The release of DAMPs to extracellular spaces is a characteristic of necrosis that distinguishes it from apoptosis[46]. Several previous studies have also reported that circulating levels of DAMPs, such as HMGB-1[47,48] or mitochondrial DNA[49] are elevated in severe COVID-19. Alveolar epithelial necrosis likely occurs very early in COVID-19 progression. The prevention of necrosis prior to the appearance of clinical symptoms is difficult; therefore, a strategy for preventing DAMPs-mediated disease aggravation is needed. Our animal study demonstrated that the inhibition of DAMPs efficiently attenuates disease progression. Further studies, including clinical trials, are warranted to investigate the clinical efficacy of DAMP inhibition in patients with COVID-19 ARDS.
In the present study, an animal model mimicking COVID-19 was established by administering the SARS-CoV-2 spike protein combined with poly (I:C). The same approach has been used in some studies[15,16,50]. Animal models treated with infectious strains of SARS-CoV-2 are often ideal models for COVID-19; however, use of infectious viruses in animal experiments can be difficult. First, SARS-CoV-2 infection does not occur in wild-type mice or rats. For infection to occur, expression of the human ACE receptor is needed. Second, facilities and equipment are needed to prevent the infection of researchers. Although the pathogenicity of SARS-CoV-2 is complex, stimulation of pathogen-associated pattern recognition receptors including toll-like receptors[26,27] and retinoic acid-inducible gene-I receptors[28] by viral components is the principal driver of lung inflammation, and subsequent alveolar tissue damage. In addition to inflammatory signatures, alveolar cell death patterns in our COVID-19 animal model were similar to those observed in human COVID-19[29,30]. Our results highlight the utility of investigating the pathophysiology and treatment of COVID-19 using animal models established with components of SARS-CoV-2.
Our data suggest that plasma M30/M65 ratio (an indicator of apoptosis in relation to total levels of epithelial cell death) is a potential marker of COVID-19 severity. Our findings agreed with a previous study that showed M30/M65 ratios of hospitalized COVID-19 patients were lower than those of non-hospitalized patients[51]. Additionally, some studies have indicated that subtypes of COVID-19 respond to treatments differently[52,53]. M30/M65 ratio may serve as a marker for selecting patients likely to benefit from anti-DAMPs treatment.
This study has some limitations. First, only patients admitted to a single center were included in the analysis due to the limited availability of clinical samples. Further studies that use samples from multiple centers several countries are warranted. Second, our animal model was created by exposing mice to components of SARS-CoV-2, and not an infectious strain of SARS-CoV-2. Despite this, the observations of a COVID-19-like pathology by the previous reports [15,16,50]and this study, support the use of the animal model. Importantly, use of a non-infectious model is convenient for laboratories that do not specialize in infectious disease research. Third, the efficacy of inhibiting a single DAMP, HMGB-1, was evaluated. Several DAMPs are released from necrotic cells; therefore, future studies that investigate whether other DAMP are promising therapeutic targets for COVID-19 are warranted.
Conclusions
In conclusion, our data indicate that necrosis, including necroptosis and pyroptosis, is the primary form of alveolar epithelial cell death in COVID-19-induced ARDS. DAMPs released from necrotic alveolar epithelial cells are potential drivers of progressive alveolar tissue damage in COVID-19; therefore, they are promising targets for preventing the aggravation of ARDS in patients with COVID-19.
Data Availability
All data produced in the present study are available upon reasonable request to the authors.
Declarations
Ethics approval and consent to participate
The study protocol was reviewed and approved by the institutional review board of Yokohama City University Hospital (approval number: B200700100, B200200048). The need for informed consent was waived by the institutional review boards because of the retrospective observational design of the study.
Consent for publication
Not applicable.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Competing interests
The authors have disclosed that they do not have any potential competing interest.
Funding
This work was partly supported by AMED under Grant Number JP20he0522001 and JSPS KAKENHI Grant Number 21K16575, and Grant for 2020-2021 Research Development Fund of Yokohama City University.
Authors’ contributions
Conceptualization and study design: KT. Acquisition of clinical study data: KT, NY, MA. Supervision of clinical sample collection: MN, IT. Acquisition of animal experiments data: KT, NT. Analysis and interpretation of data: KT, NY, NT, TM, TG. Manuscript Writing: KT. Manuscript Editing: All investigators.
Acknowledgements
We would like to thank Ms. Yuki Yuba (Department of Anesthesiology and Critical Care Medicine) for technical assistance. We also thank Department of Emergency Medicine, Department of Microbiology, and Yokohama City University Center for Novel and Exploratory Clinical Trials for collecting and providing the blood samples. This work was partly supported by AMED under Grant Number JP20he0522001 and JSPS KAKENHI Grant Number 21K16575, and Grant for 2020-2021 Research Development Fund of Yokohama City University.
Footnotes
We have added the analysis of bronchalveolar fluids of patients with COVID-19-induced ARDS.
List of abbreviations
- ARDS
- Acute respiratory distress syndrome
- ANG
- angiopoietin
- BALF
- bronchoalveolar lavage fluid
- COVID-19
- coronavirus disease 2019
- CK
- cytokeratin
- DAMPs
- damage-associated molecular patterns
- ELISA
- enzyme-linked immunosorbent assay
- HMGB
- high mobility group box
- LPS
- lipopolysaccharide
- PVDF
- polyvinylidene fluoride
- sRAGE
- receptors for advanced glycation end products
- SARS-CoV-2
- severe acute respiratory syndrome coronavirus 2
- SP
- surfactant protein
References



