
Abstract
Type 2 diabetes mellitus (T2D) is one of the most prevalent diseases in the world and presents a major health and economic burden, a notable proportion of which could be alleviated with improved early prediction and intervention. While standard risk factors including age, obesity, and hypertension have shown good predictive performance, we show that the use of CpG DNA methylation information leads to a significant improvement in the prediction of 10-year T2D incidence risk.
Whilst previous studies have been largely constrained by linear assumptions and the use of CpGs one-at-the-time, we have adopted a more flexible approach based on a range of linear and tree-ensemble models for classification and time-to-event prediction. Using the Generation Scotland cohort (n=9,537) our best performing model (Area Under the Curve (AUC)=0.880, Precision Recall AUC (PRAUC)=0.539, McFadden’s R2=0.316) used a LASSO Cox proportional-hazards predictor and showed notable improvement in onset prediction, above and beyond standard risk factors (AUC=0.860, PRAUC=0.444 R2=0.261). Replication of the main finding was observed in an external test dataset (the German-based KORA study, p=3.7×10−4). Tree-ensemble methods provided comparable performance and future improvements to these models are discussed.
Finally, we introduce MethylPipeR, an R package with accompanying user interface, for systematic and reproducible development of complex trait and incident disease predictors. While MethylPipeR was applied to incident T2D prediction with DNA methylation in our experiments, the package is designed for generalised development of predictive models and is applicable to a wide range of omics data and target traits.
Introduction
Diabetes mellitus is one of the most prevalent diseases in the world and a leading cause of mortality. Around half a billion people live with diabetes worldwide, with type 2 diabetes (T2D) making up about 90% of these cases [1]. Individuals with diabetes can suffer from debilitating complications including nerve damage, kidney disease and blindness [2]. The disease also increases the future risk of dementia and cardiovascular disease [3], with recent studies highlighting obesity and T2D as risk factors for COVID-19 disease severity and ICU admission [4]. Furthermore, risk of complications increases over time and is exacerbated if blood-glucose levels are poorly managed. Despite developments in the way T2D can be managed for patients, these treatments are reactive, focusing on patients that have already been diagnosed. Early prevention and detection could therefore have major health and economic impacts.
While the mechanisms of insulin resistance in T2D are well-known, the interaction between genetic and environmental factors that increase T2D susceptibility are less understood. Epigenetics is the study of heritable changes to DNA that do not modify its nucleotide sequence. A commonly studied form of this is DNA methylation (DNAm), whereby methyl groups are attached to the DNA molecule - most commonly to the 5-carbon on a cytosine in a cytosine-guanine pair (CpG). Due to its involvement with gene expression and gene- environment interactions, DNAm can provide dynamic predictive information for disease risk for an individual. For example, penalised regression models have been used to show that weighted linear CpG predictors can explain a substantial proportion of phenotypic variance of modifiable health factors including body mass index (BMI) (12.5%), HDL cholesterol (15.6%) and smoking status (60.9%) [5].
Epigenome-wide association studies (EWAS) have identified a number of CpG sites significantly associated with T2D [6-10] as well as related risk factors such as cardiovascular disease [11] and obesity [12, 13]. While these provide some predictive performance for T2D prevalence, incident T2D has been less well studied. Given that preventative lifestyle changes have been shown to effectively reduce T2D onset [14], prediction of T2D incidence years ahead of time would be greatly beneficial in stratifying populations so those at high risk can be monitored and treated with early interventions.
Currently, most studies generating DNAm predictors consider marginal CpG effects or assume only linear additive effects between CpGs. The use of predictive models that can incorporate both interaction and non-linear effects could capture more complex relationships between variables, resulting in greater prediction accuracy.
Here, we use one of the world’s largest studies with paired genome-wide DNAm and data linkage to electronic health records (EHR), Generation Scotland (n=9,537, n=428 incident T2D cases over 14 years of follow-up), to train and test epigenetic scores (EpiScores) for T2D. Availability of time-to-event information (time from baseline, here defined as DNAm sampling date, to disease onset or censoring) enables the use of survival models. We consider penalised linear models and tree-ensemble models in both classification and survival/time-to- event model forms and describe the added contribution of these DNAm predictors over and above standard risk factors e.g. age, sex and BMI. We then validate the best performing model in the KORA S4 cohort [15], providing further evidence of the applicability of our EpiScore to external populations.
As T2D and related risk factors such as obesity have also been associated with severity of COVID-19 infection, we also evaluate the performance of T2D EpiScores on predicting long COVID-19 and hospitalisation in infected individuals in the Generation Scotland study.
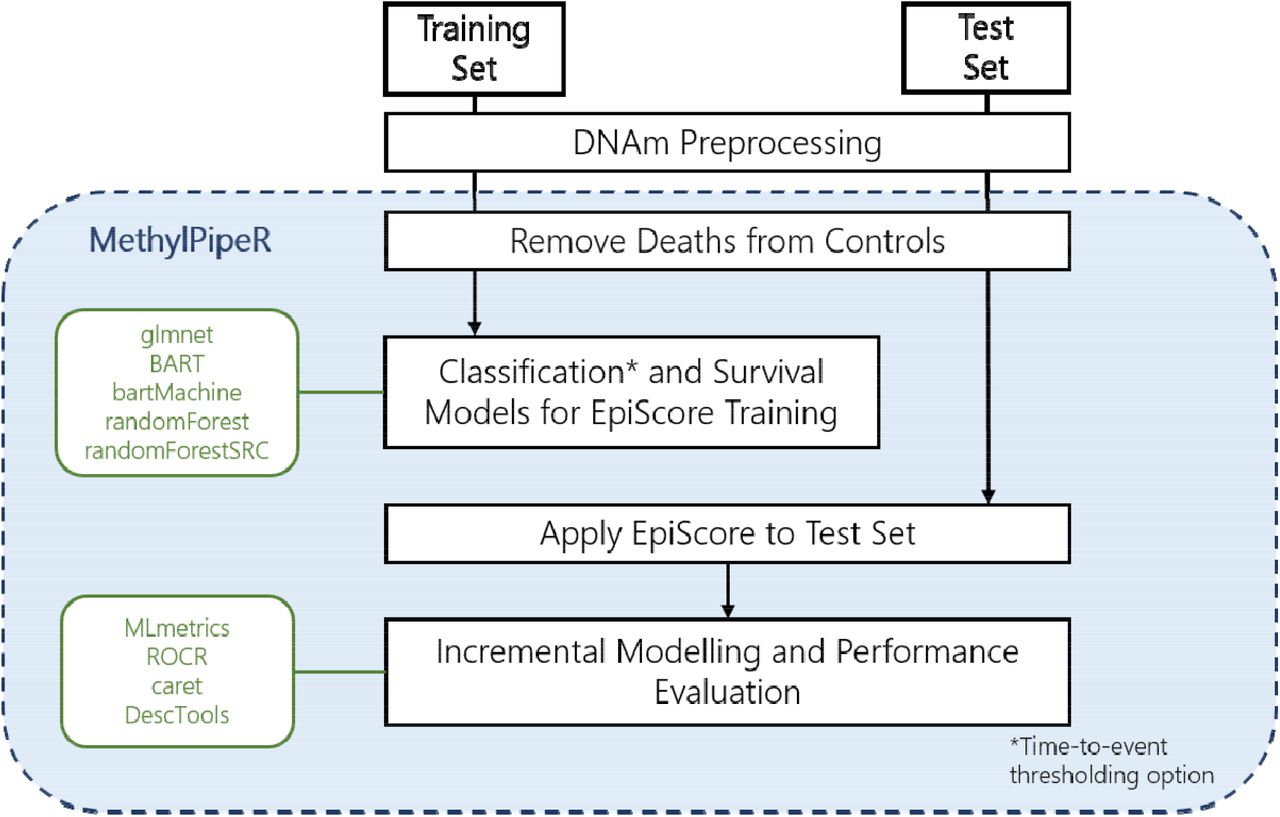
The analysis pipeline is implemented via a new R package, MethylPipeR, along with accompanying user interface, for systematic and reproducible development of complex trait and incident disease predictors. MethylPipeR provides functionality for tasks such as model fitting, prediction and performance evaluation as well as automatic logging of experiments and trained models. This is complemented by MethylPipeR-UI which provides an interface to the R package functionality while removing the need to write scripts. While MethylPipeR was applied to incident T2D prediction with DNA methylation in our experiments, the package is designed for generalised development of predictive models and is applicable to a wide range of omics data and target traits. MethylPipeR and MethlyPipeR-UI are publicly available at https://github.com/marioni-group/MethylPipeR and https://github.com/marioni-group/MethylPipeR-UI respectively. Supplementary Figure 1 shows an example from the MethylPipeR-UI interface including functionality such as data upload, specification of model options and visualisation of model diagnostics.
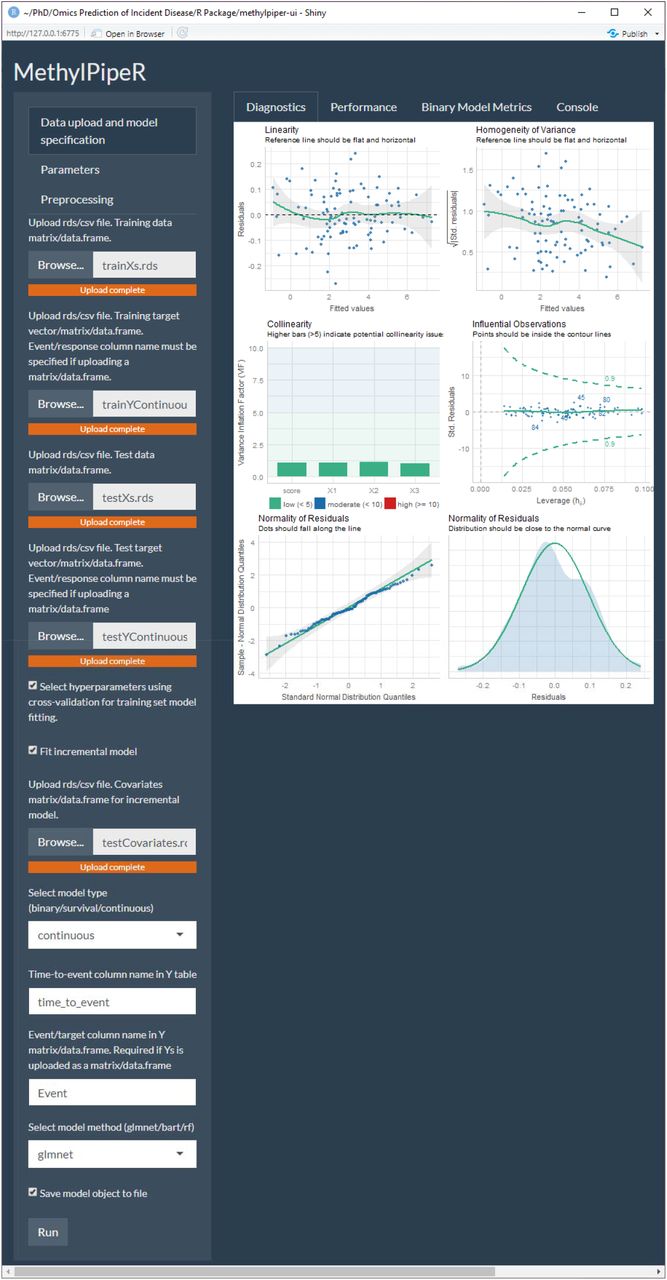
An example from the MethylPipeR-UI Shiny app.
Methods
Generation Scotland
DNAm and linked health data were obtained from Generation Scotland [16], a family- structured population-based cohort. The cohort consists of 23,960 participants aged 18-99 years at recruitment (between 2006 and 2011), of whom 9,537 currently have genome-wide DNAm data available (Illumina EPIC array) following quality control. DNAm quality control consisted of removing probes with outliers, low bead count in ≥5% of samples or a high detection p-value (>0.05) in more than 5% of samples. Samples with mismatch between predicted and recorded sex or ≥ 1% of CpGs with detection p-value > 0.05 were also removed. To enable the predictors to be applied to existing cohort studies with older Illumina array data, CpGs were filtered to the intersection of the 450k and EPIC array sites (n=398,422 CpGs).
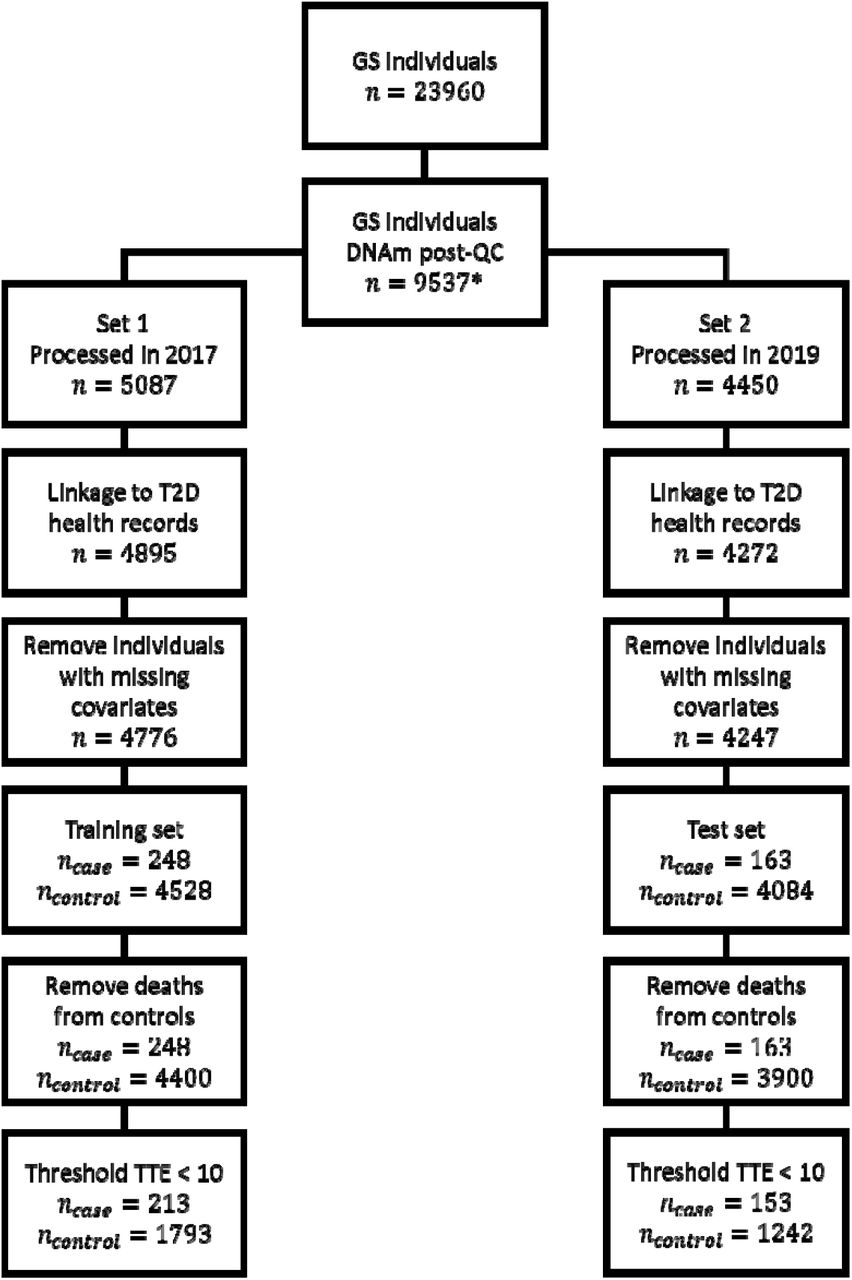
DNAm data were processed as two separate sets, with 5,087 (Set 1) and 4,450 (Set 2) individuals. Processing took place in 2017 and 2019, respectively. Set 1 included related individuals while all individuals in Set 2 were unrelated to each other and to individuals in Set 1 (genetic relationship matrix (GRM) threshold <0.05). In our experiments, Set 2 was used as the training set and Set 1 as the test set to avoid the presence of related individuals in the training set.
Participant health measures such as age, body mass index (BMI) and sex were taken at baseline as well as self-reported hypertension and family (parent or sibling) history of T2D. BMI was calculated as the individual’s weight in kg divided by the square of their height in metres. Missing values in the health measures were treated as missing-completely-at-random and the corresponding individuals were excluded (nSet 1=119, nSet 2=25).
Disease cases were ascertained through data linkage to NHS Scotland health records consisting of hospital (ICD codes) and GP records (Read2 codes). Prevalent cases were identified from a baseline questionnaire (self-reported) or from ICD/Read2 codes dated prior to baseline and removed from the dataset. Type 1 and juvenile cases were treated as control observations. All included and excluded terms are listed in Supplementary Table 1. A total of 428 incident cases were observed over the follow up period (from recruitment date to 09/2020): 80% of cases had a C10F. “type 2 diabetes mellitus” code; 14% had a C10.. “diabetes mellitus” code; 5% had a C109. “Non-insulin dependent diabetes mellitus” code. The remaining codes diabetes mellitus with: neurological manifestation (n=1); renal manifestation (n=4); and with adult onset and no mention of complication (n=3).
Outcome Definition for 10-Year Onset Prediction
Linkage to NHS Scotland health records provided dates for disease diagnoses from which age-at-onset was calculated. Along with age at baseline (DNAm sampling), these were used to calculate the time-to-event, measured in years, for each individual. For incident T2D cases and controls, time-to-event was defined as the time from baseline to disease onset and censoring (end of follow up period or died without a disease diagnosis), respectively.
Our primary prediction outcome was incident T2D diagnosis within 10 years. For this purpose, two types of model were used. For binary classification models, further preprocessing of cases and controls was performed to reflect 10-year onset prediction. Incident cases with time-to-event >10 years were treated as controls (training n=10, test n=35). Controls with time-to-censoring ≤10 years were excluded (training set n=2,668, test n=2,642), given it was unknown if those individuals would develop T2D within the 10-year period.
For survival models, this further preprocessing was only applied to the test set due to the ability to incorporate censoring information in the training set. For these models, controls that had died within the 10 year period were excluded from the training set (n=129) as it was unknown if the death was due to a diabetes-related (and therefore confounding) risk factor.
There were 153 cases and 1,242 controls in the training set and 213 cases and 1,793 controls in the test set. The numbers of individuals/cases and controls after each preprocessing step are also shown in Supplementary Figure 2.

Preprocessing steps for Generation Scotland with number of individuals/cases and controls after each step.
Incremental R2 Modelling
In the test set, a null model was defined via logistic regression of 10-year risk of diabetes with age, sex, BMI, self-reported hypertension, and self-reported family (sibling or parental) history of diabetes as predictors. The area under the curve (AUC), area under the precision- recall curve (PRAUC) and the adjusted McFadden’s pseudo-R2 (henceforth referred to as R2) for the model were considered as classification/fit metrics. DNAm predictors were then generated in the training dataset using a variety of machine learning methods, via the MethylPipeR package (Figure 1), before being applied to the test set in an incremental R2 modelling approach (further detail in Supplementary Methods).

The prediction pipeline and functionality provided in MethylPipeR
Penalised regression predictors
Since the number of CpGs (n=398,422) was much greater than the number of rows in the training set (n=1,395 after preprocessing), a regularisation method was required to reduce overfitting of the logistic and Cox proportional hazards regression models.
Lasso, elastic-net and ridge penalisation were fit to the training set DNAm data using glmnet [17, 18] with the best shrinkage parameter (λ) chosen by cross-validation. For elastic-net models, α = 0.5 was used for the L1, L2 mixing parameter (full details in Supplementary methods). Models with and without weights to correct for an imbalance in the numbers of cases compared to controls were also considered.
Tree Ensemble Models
Due to computational limitations and probable overfitting in using the tree ensemble models on all CpGs in the dataset, variable pre-selection was based on the coefficients in the penalised logistic models. Each tree-ensemble model was evaluated with the features corresponding to non-zero coefficients from the logistic lasso model and also with those selected by elastic-net.
Two tree ensemble approaches were used: random forest and Bayesian Additive Regression Trees (BART). Random forest [19] is an ensemble machine learning model that estimates a function by averaging the output from a set of independently trained decision trees. During model fitting, each tree is built using a different subset of the variables from the training set to prevent individual trees from overfitting to the whole dataset. BART is a nonparametric method that estimates a function as a sum over a set of regression trees. BART incorporates the ability to model both additive and interaction effects and has shown high predictive performance in comparison with similar methods [20]. In addition to binary classification, survival random forest [21] and survival BART [22] models were considered.
Evaluating Predictive Performance
AUC and PRAUC were calculated as measures of predictive performance as the discrimination threshold was varied. PRAUC is more informative in situations where there is a class imbalance in the test set. R2 was evaluated for each model with the incremental R2 calculated as the difference in R2 between the null model and the full model. Additionally, binary classification metrics consisting of sensitivity (recall), specificity, positive predictive value (PPV/precision) and negative predictive value (NPV) were calculated. These metrics require selection of a probability threshold to assign positive/negative class predictions and have varying behaviour as this threshold is altered. Therefore, each of the metrics were calculated at a range of thresholds between 0-1 in increments of 0.01.
Model calibration was examined by comparing predicted probabilities with actual case/control proportions. The test data was sorted by predicted probability and divided into bins; the mean predicted probability and the proportion of cases was calculated for each bin.
Selected-CpG Comparison with EWAS Catalog
Lasso and elastic-net model penalties result in most coefficients being set to 0, effectively selecting a small subset of CpGs and covariates to be used in prediction. The selected CpGs for the highest R2 penalised model were queried in the EWAS Catalog [23] to identify traits that have previously been linked to these sites at an epigenome-wide significance threshold of P < 3.6 × 10−8 in studies with a sample size > 1,000 [24].
Validation in KORA S4
The highest R2 model (weighted Cox lasso) was applied to the KORA S4 cohort [15]. This cohort consisted of 1,451 individuals in southern Germany, aged 25-74. Full summary statistics are shown in Supplementary Table 2. Similar to the approach in the Generation Scotland test set, an EpiScore was computed for each individual in the KORA dataset. Evaluation was then performed in incremental R2 approach. Additional cohort and methods details are provided in Supplementary methods.
EpiScore Prediction of Long COVID-19/Hospitalisation
The subset of the Generation Scotland cohort with reported COVID-19 infection (positive test or suspected) in the CovidLife study [25] were used for prediction of long COVID-19 and hospitalisation from COVID-19. Long COVID-19 cases were defined here as individuals with self-reported symptoms lasting ≥ 4 weeks. Hospitalisation cases were defined as hospital admissions with accompanying ICD10 codes U07.1 (confirmed COVID-19 test) and U07.2 (clinically diagnosed), derived from the Scottish Morbidity Records (SMR01). Details of the method and summary statistics are shown in Supplementary methods and Supplementary Table 3.
Results
After preprocessing, the mean time-to-onset of T2D was 5.24 and 5.12 years for the training (n=153 cases) and test (n=213 cases) sets, respectively. Mean age-at-onset was also similar between the training and test set at 62.1 and 59.8 years and the mean BMI for cases (at baseline) was 31.8 and 32.7. The full set of summary statistics for cases and controls in both sets are shown in Table 1 and Supplementary Table 4. The machine learning prediction pipeline of the MethylPipeR package is shown in Figure 1.
Summary information for the Generation Scotland training and test sets.* Summary information is mean (SD) or N (%).
Null Model for the Incremental Modelling Approach
A logistic regression model in the test set with age, sex, BMI, self-reported hypertension, and family history of diabetes as predictors yielded good classification metrics: AUC=0.860, PRAUC=0.444, R2=0.261.
Penalised Logistic Regression and Cox PH
Lasso and elastic-net models showed similar values across all metrics with test set AUC, PRAUC and R2 ranges of 0.875-0.878, 0.490-0.500 and 0.294-0.301, respectively (Supplementary Table 5). Logistic ridge offered minimal improvement over the null model and the weighted logistic ridge was not included in the results due to computational issues in model fitting.
Compared to the logistic models, the Cox models showed a greater difference in performance metrics with test set AUC, PRAUC and R2 ranges of 0.870-0.881, 0.483-0.539 and 0.285- 0.316, respectively (Supplementary Table 5). This difference was most apparent when comparing the weighted vs. non-weighted cox models; for example, adding class weights for Cox elastic-net led to an increase of 1.1%, 5.2% and 3.1% in absolute terms for AUC, PRAUC and R2.
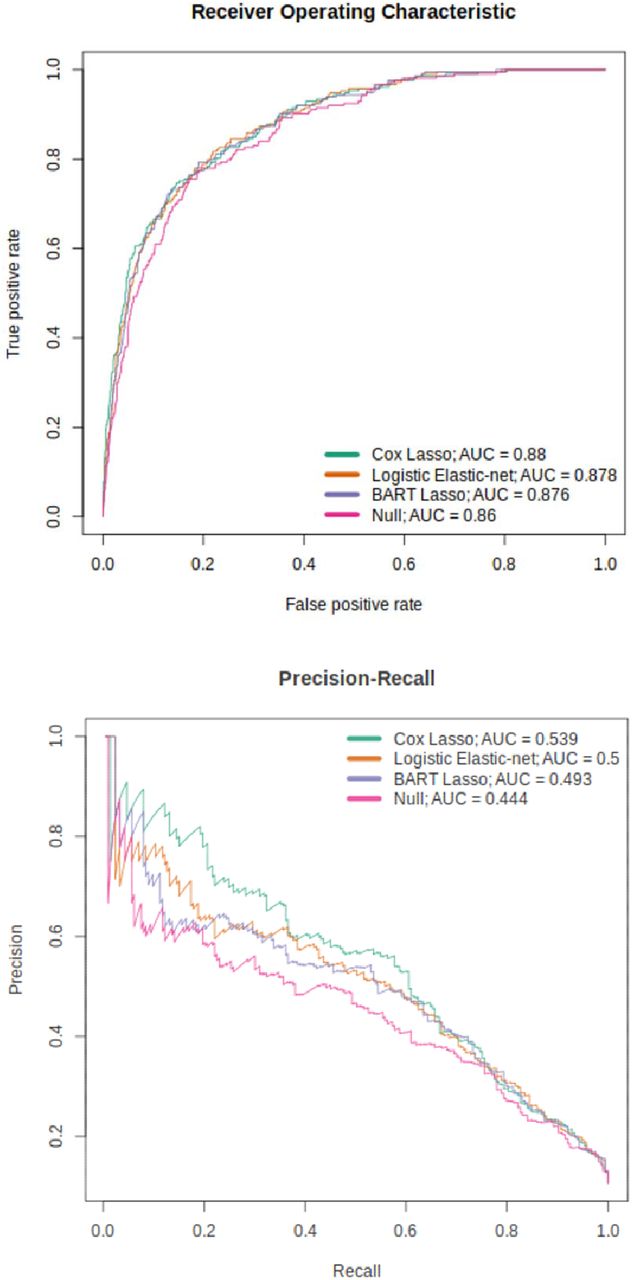
The best-performing Cox and logistic models are presented in Table 2 and Figure 2; the CpG weights for these models are shown in Supplementary Tables 6 and 7. Comparing all models together (Supplementary Table 5), the weighted Cox models outperform all others. Cox lasso (with weights) showed the highest PRAUC and R2 - 0.539 and 0.316, respectively - and an AUC of 0.880, 0.1% short of the highest AUC of 0.881 (corresponding to Cox elastic- net with weights). The effect of class weights was further emphasised here as the higher performance of the Cox predictors above the logistic predictors was only present when weights were applied. All models other than the logistic ridge showed a p-value < 0.05 for the EpiScore coefficient in the incremental R2 full model, with a range of 1.71×10−16 to 3.15×10−4.
Logistic regression metrics for 10-year prediction of diabetes in the test set (best Cox, Logistic, BART, and Random Forest predictor output). AUC = Area Under the Curve; PRAUC = Precision Recall AUC; R2 = McFadden’s pseudo-R2; EpiScore = Epigenetic Score.
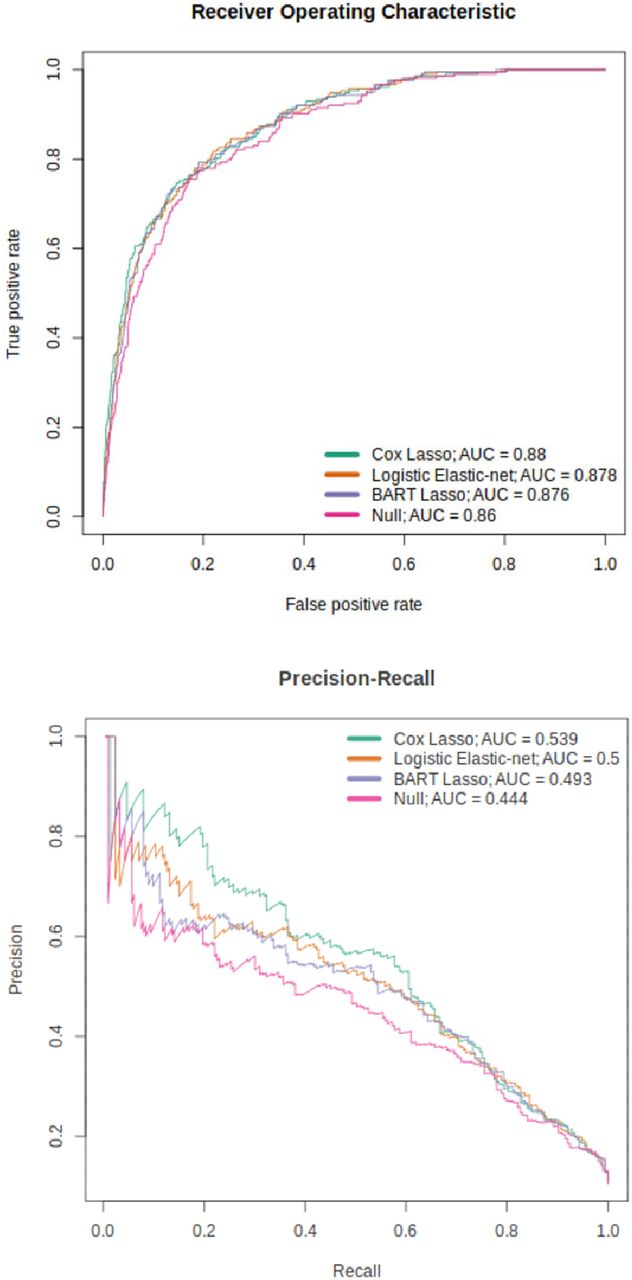
ROC and PR curves for the top-performing Cox, logistic and tree-ensemble models (weighted Cox lasso, weighted logistic elastic-net and BART with weighted lasso features).
Tree-Ensemble Models
Overall, the tree-ensemble models resulted in lower but comparable results to logistic and Cox models. AUC, PRAUC and R2 values showed ranges of 0.865-0.876, 0.458-493 and 0.269 and 0.293, respectively (Supplementary Table 5). With the exception of the survival random forest with weighted lasso features (see Supplementary Methods for details), the BART models outperformed the random forest methods in both survival and classification formulations. In addition, with the exception of BART with elastic-net features, models that used weighted logistic model-selected features achieved higher performance than those using unweighted logistic features.
Differences in performance between survival and classification formulations of the same model type were relatively small. For instance, the best performing tree-ensemble model, classification BART (with weighted lasso features) showed AUC, PRAUC and R2 values of 0.876, 0.493 and 0.293 respectively. Similarly, survival BART (with weighted lasso features) gave values of 0.874, 0.488 and 0.288, corresponding to differences of 0.002, 0.005 and 0.005 across the respective metrics. The best-performing BART and random forest models are shown in Table 2 with the BART model also highlighted in Figure 2.
Binary Classification Metrics and Model Calibration
Figure 3. shows how sensitivity, specificity, PPV and NPV vary for the best performing logistic model (weighted logistic elastic-net) and tree-ensemble model (BART with weighted lasso features). These are shown for the EpiScore applied to the test set without additional covariates. In general, as the classification probability threshold is increased, sensitivity and NPV decrease while specificity increases. However, there are clear differences between the two; for example: the logistic model shows greater PPV values in the high probability threshold range, while the same metric drops off in BART. In addition, while the overall trends in sensitivity, specificity and NPV are similar, the rate of change across the probability thresholds differ. BART shows greater changes in these metrics in the low probability threshold range. The differences in the two models is also apparent from the calibration curves (Supplementary Figure 3). Additionally, the effects of these differences on the number of true positives and true negatives are illustrated in Figure 4.
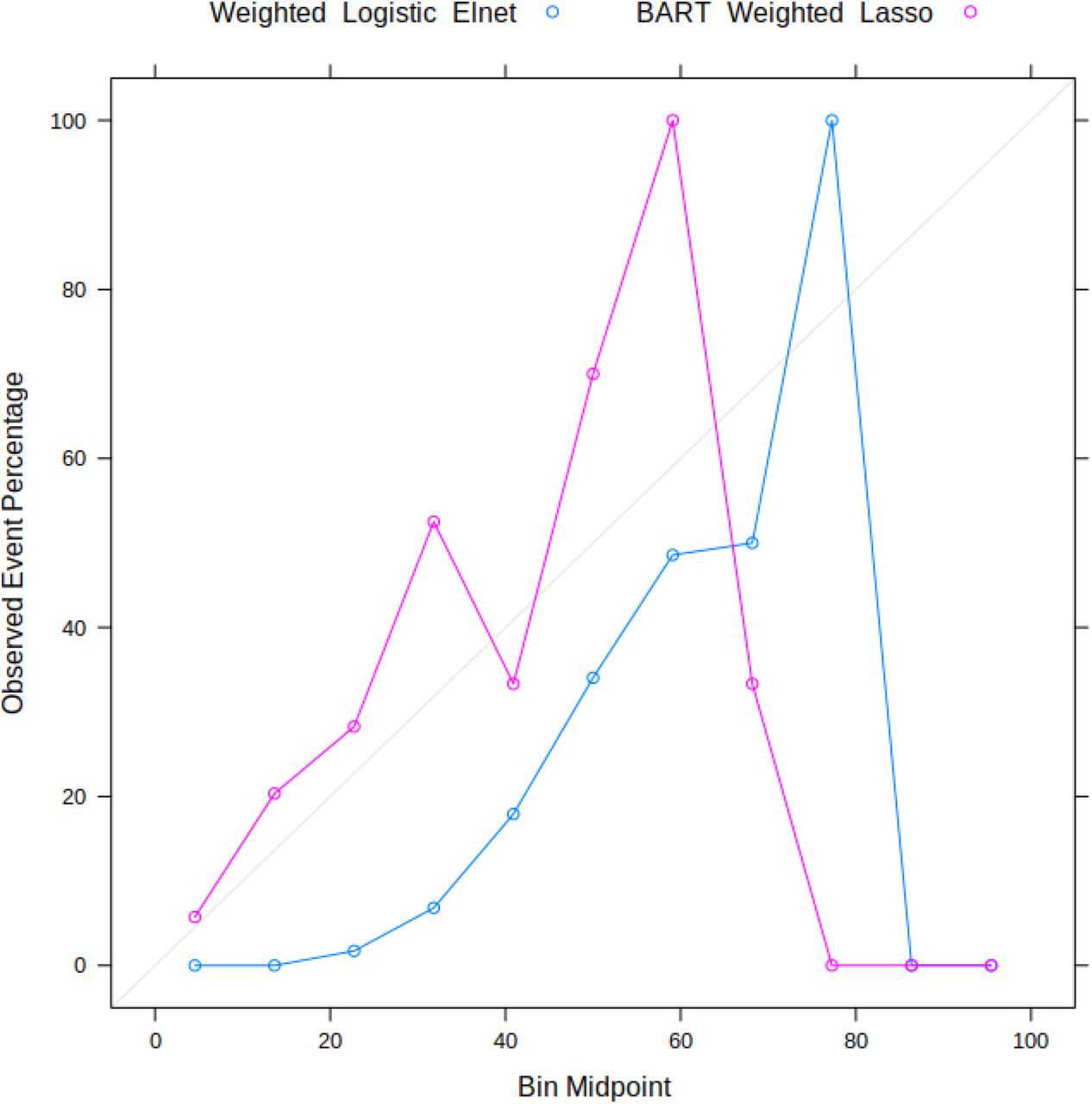
Calibration plots for the EpiScore generated from the weighted logistic elastic-net model and BART with weighted lasso-selected features.

Classification metrics (sensitivity, specificity, PPV and NPV) for the EpiScores generated from the weighted logistic elastic-net model (top) and BART with weighted lasso- selected features (bottom). The difference in PPV behaviour in the high threshold ranges between the two models is due to the low number of individuals with an EpiScore in those ranges.

Confusion matrix plot of true/false positives/negatives for the null model and full model in the Generation Scotland test dataset. (Full model uses EpiScore from Cox Lasso with weights.) The table shows binary classification metrics at a probability threshold of 0.5 (corresponding to the vertical green line in each plot).
Selected CpGs
The weighted Cox lasso model assigned non-zero coefficients to 69 CpGs (Supplementary Table 6). After filtering the EWAS Catalog by p-value (P<3.6×10−8) and sample size (N>1,000), 20 of the model-selected CpGs were present in the catalog. These CpGs corresponded to 176 entries and showed epigenome-wide associations with traits including: serum HDL cholesterol, serum triglycerides, smoking, C-reactive protein, BMI and age (Supplementary Table 8).
Validation in KORA S4
Prediction of incident diabetes in the KORA S4 cohort using the weighted Cox lasso model showed good replication of EpiScore performance (P=3.7×10−4) with increases of 0.93%, 1.9% and 1.2% in absolute terms above the null model values for AUC, PRAUC and R2 respectively. Further details are provided in Supplementary Table 9.
EpiScore Prediction of Long COVID-19/Hospitalisation
Age at COVID-19 diagnosis and sex were not predictive of long COVID-19 in a logistic regression model (Supplementary Table 10). In addition, the weighted Cox Lasso T2D EpiScore (from baseline blood-based DNA collected between 2006 and 2011) did not show a significant improvement in prediction when added as a variable. Similarly, the EpiScore was not predictive of hospitalisation after COVID-19 infection.
Discussion
Utilising a large cohort with genome-wide epigenetic data and health records linkage to longitudinal primary and secondary care health diagnoses, we have shown that DNAm-based predictors augment standard risk factors in the prediction of incident type 2 diabetes. The best model with traditional risk factors yielded an AUC of 0.860 compared to 0.881 when DNAm was also considered. PRAUC increased from 0.444 to 0.539 and R2 from 0.261 to 0.316. Using a variety of linear and non-linear models, we showed that overall, weighted penalised Cox PH models produced the most predictive EpiScore. This EpiScore also showed good external validation performance in the KORA S4 cohort. Beyond the T2D analysis presented here, we have developed the MethylPipeR R package to facilitate reproducible machine learning time-to-event and binary prediction using DNAm or other types of high-dimensional omics data.
Determining a ‘best’ model is complicated and depends on the trade-off that a user wishes to make. Here, we optimised AUC, PRAUC and R2 but binary classification metrics vary by method and classification threshold. When using classifiers in clinical settings, decisions need to be made about the number of patients that can be recommended for intervention as well as the acceptable proportion of false positives and false negatives. In addition, an assessment of calibration is also critical [26]. Investigation of how these related criteria could assist in deciding an optimal threshold given clinical constraints and provide a more comprehensive assessment of model predictions than AUCs or metrics at the commonly- utilised threshold of 0.5.
Analysis of the EpiScores with COVID-19 phenotypes showed that while the scores considerably increased incident T2D prediction performance, they are not indicative of susceptibility to severe COVID-19 symptoms, despite previously found associations between the two [4].
Several CpGs from the EpiScores were previously identified as epigenome-wide significant correlates of traits commonly linked to T2D [10,13, 27-31]. Future work could investigate overlap between these and time-to-event EWAS studies. Further studies could also include DNAm predictors for traditional risk factors, such as BMI [5], and protein EpiScores known to be linked to T2D and related pathways [32].
Limitations include the relatively small number of disease cases in the dataset, the limited hyperparameter optimisation performed for BART and the relatively simple variable pre- selection method for tree-ensemble methods. Given the lower but competitive performance of these methods compared to the best models in this study, there is potential for additional improvement in predictive performance with further investigation of more advanced pre- selection. This is particularly important when we consider that the pre-selection step utilised linear models prior to the non-linear model fitting. The model fitting and pre-selection were also performed using the same training set which may have introduced selection bias [33]. In addition, factors such as overfitting, related individuals in the test set and batch effects between the two rounds of DNAm data processing may all have an effect on test-set AUC. Finally, a small proportion of the linkage codes used to define diabetes included broad terms that were non-specific to T2D; however, late age of onset in these individuals meant there was a high likelihood that they had developed T2D. EpiScores for T2D-associated proteins have also been shown to replicate incident T2D-protein associations within this sample [32] suggesting that the case definitions we use capture biological signals relevant to T2D.
There are numerous strengths to our study. Firstly, the models used capture relationships between CpGs as well as time-to-event information, which is not possible using traditional EWAS methods. Secondly, data linkage to health care measures provided comprehensive T2D incidence data in a very large cohort study, Generation Scotland. Validation performance in the KORA cohort also strengthened evidence for the applicability of the models to other populations. Finally, the R package, MethylPipeR, encourages reproducibility and allows others to develop similar predictors on new data with minimal setup.
In conclusion, we have demonstrated the potential for DNA methylation data to provide notable improvement in predictive performance for incident T2D, as compared to traditional risk factors (age, sex, BMI, hypertension, and family history). We evaluated a wide range of models with a systematic approach and presented a framework with the ability to generalise to other traits and datasets for training and testing predictors in future studies.
Data Availability
According to the terms of consent for Generation Scotland participants, access to data must be reviewed by the Generation Scotland Access Committee. Applications should be made to access@generationscotland.org. All code is available with open access at the following Gitlab repository: https://github.com/marioni-group MethylPipeR (version 1.0.0) is available at: https://github.com/marioni-group/MethylPipeR The informed consents given by KORA study participants do not cover data posting in public databases. However, data are available upon request from KORA Project Application Self-Service Tool (https://epi.helmholtz-muenchen.de/). Data requests can be submitted online and are subject to approval by the KORA Board.
Declarations
Ethics approval and consent to participate
All components of Generation Scotland received ethical approval from the NHS Tayside Committee on Medical Research Ethics (REC Reference Number: 05/S1401/89). Generation Scotland has also been granted Research Tissue Bank status by the East of Scotland Research Ethics Service (REC Reference Number: 20-ES-0021), providing generic ethical approval for a wide range of uses within medical research.
The KORA studies were approved by the Ethics Committee of the Bavarian Medical Association (Bayerische Landesärztekammer; S4: #99186) and were conducted according to the principles expressed in the Declaration of Helsinki. All study participants gave their written informed consent.
Availability of Data and Material
According to the terms of consent for Generation Scotland participants, access to data must be reviewed by the Generation Scotland Access Committee. Applications should be made to access@generationscotland.org.
All code is available with open access at the following Gitlab repository: https://github.com/marioni-group
MethylPipeR (version 1.0.0) is available at: https://github.com/marioni-group/MethylPipeRThe informed consents given by KORA study participants do not cover data posting in public databases. However, data are available upon request from KORA Project Application Self- Service Tool (https://epi.helmholtz-muenchen.de/). Data requests can be submitted online and are subject to approval by the KORA Board.
Competing Interests
R.E.M has received a speaker fee from Illumina and is an advisor to the Epigenetic Clock Development Foundation. A.M.M has previously received speaker fees from Janssen and Illumina and research funding from The Sackler Trust. All other authors declare no competing interests.
Supplementary Methods
Methylation Risk Scores and Incremental R2
Each model was fit to the training set using the CpGs as features. This trained model was used to create an Epigenetic Score (EpiScore) for each individual in the test set and its predictive performance was evaluated by fitting a logistic model (full model) on the test set including the EpiScore and covariates (age, sex, BMI, hypertension and parent/sibling T2D). This was compared to the null model (logistic with covariates only) to assess the difference in metrics such as the area under the curve (AUC), area under the precision-recall curve (PRAUC) and the adjusted McFadden’s pseudo-R2.
Penalised Logistic Regression
Since the number of features (=398,422) was much greater than the number of individuals in the training set (=1,395 after data preprocessing), a regularisation method was required to reduce overfitting of the logistic regression models.
Logistic regression models with lasso, elastic-net and ridge penalisation were fit to the training set using glmnet (version 4.1-1) [17, 18] with the best λ chosen by cross-validation, corresponding to the minimum mean cross-validated error. For elastic-net models, α = 0.5 was used for the L1, L2 mixing parameter.
Hyperparameter optimisation and cross validation (CV) were used to estimate an optimal value of λ for each logistic regression model. The training set was divided equally into three partitions. For each pre-selected value of λ, three models were fit, each using two of the partitions as the training set and the third for prediction. The mean binomial deviance over the three models was then calculated. The model using the λ that minimised this was chosen to evaluate on the test set. Three-fold CV was used to balance the advantages provided by using a greater number of folds with the limitations of the number of cases in each fold as well as required computation time. In addition, the folds were designed to include an equal number of cases to avoid folds with few or zero cases.
Penalised Cox Proportional-hazards Survival Model
To evaluate the benefit of incorporating time-to-event information, penalised Cox proportional-hazards (Cox PH) models were compared with the penalised logistic regression models. Cox PH models with lasso and elastic-net regularisation were used to infer the hazard function parameters. The resulting linear predictor for the hazard function was used to generate an EpiScore for each individual in the test set. Optimal values of λ were estimated using the same CV method as in the logistic regression models, except mean partial likelihood was used as the metric for deciding the best model. Similar to the penalised logistic models, the penalised Cox PH models were fit using glmnet.
Random Forest
Random forest [19] is an ensemble machine learning model that estimates a function by averaging the output from a set of independently trained decision trees. During model fitting, each tree is built using a different subset of the variables and the training set to prevent individual trees from overfitting to the whole dataset. In addition, random survival forests adapts the original method to incorporate right-censored time-to-event data [21]. In this study, both random forest for classification and survival random forest were applied to compare their difference in predictive performance.
The hyperparameters corresponding to the number of trees (ntrees), the number of variables considered at each tree split (mtry) and the minimum terminal node size (nodesize) were selected using a grid-search CV method. Supplementary Table 11 shows the set of values that were tried for each hyperparameter. The R packages randomForest (version 4.6-14) [34] and randomForestSRC (version 2.11.0) [35] were used for classification and survival random forest models respectively.
Bayesian Additive Regression Trees
Bayesian Additive Regression Trees (BART) [20] is a nonparametric method that estimates a function as a sum over a set of regression trees. BART incorporates the ability to model both additive and interaction effects and has shown high predictive performance in comparison with similar methods. To reduce overfitting and model uncertainty in parameters and predictions, BART uses prior distributions over tree-related parameters. Posterior estimates are obtained in a Bayesian framework through Markov Chain Monte Carlo (MCMC).
For classification BART, 20,000 posterior samples for model parameters were kept after 10,000 burn-in samples and the mean probability from was used as the model output.
A variant of BART for survival analysis [22] was also used for 10-year onset prediction. 1,000 posterior samples for model parameters were kept after 500 burn-in samples. These were used to generate 10-year survival probabilities on the test set. This resulted in 1,000 survival probabilities for each individual in the test set, the mean of which was used as their survival prediction. The 10-year onset probability was taken as 1 - 10-year survival probability.
Due to the computation time requirements of MCMC sampling and the apparent robustness of BART to hyperparameter misspecification [20], all BART models (classification and survival) were run with 100 trees and remaining hyperparameters set to default. The R packages bartMachine (version 1.2.6) [36] and BART (version 2.9) [37] were used for classification and survival BART respectively.
Weights for Class Imbalance
Both Set 1 and Set 2 showed a high ratio of controls to cases. When a high imbalance between negatives and positives is present, weights are often applied to the data to address under-representation of the positive class. To evaluate their effectiveness, all penalised models (logistic and Cox PH) were also run with and without class weights. Cases were given a higher weighting in the maximum likelihood estimate to account for their low proportion in the training set. These were calculated using the following heuristic inspired by [38]:
 where ntotal is the number of elements in the training set, wcase is the weight applied to each case and ncase is the number of cases in the training set (similar for controls). Using these weights ensured that half the weighting was applied to cases and similarly for controls. In addition, the sum of all weights = ntotal. To assess the impact of pre-selection on tree- ensemble methods, each was evaluated using variables selected using the penalised logistic models both with and without weights applied.
where ntotal is the number of elements in the training set, wcase is the weight applied to each case and ncase is the number of cases in the training set (similar for controls). Using these weights ensured that half the weighting was applied to cases and similarly for controls. In addition, the sum of all weights = ntotal. To assess the impact of pre-selection on tree- ensemble methods, each was evaluated using variables selected using the penalised logistic models both with and without weights applied.
Validation of Best Performing Model in KORA S4
The present analyses are based on a subsample of the participants of the KORA S4 study. KORA (Cooperative Health Research in the Region of Augsburg) is a research platform performing population-based surveys and subsequent follow-ups in the region of Augsburg in Southern Germany [39]. Participants were aged between 25-74 years and each completed a health questionnaire, providing details on health status and medication. Blood samples were also taken for assaying of omics data. This study used a subsample of the 1,451 participants of the KORA S4 study with DNAm and incident T2D data available.
The best performing model selected for the Generation Scotland cohort (weighted Cox lasso) was used for prediction of incident T2D in the KORA S4 cohort.
For diabetes morbidity, the data are limited the follow-up to 10 years - starting from KORA S4. For incident T2D all prevalent diabetics as well all other diabetes types except T2D cases are excluded. Age, body mass index (BMI), hypertension, sex as well as self-reported family (mother or father) history of T2D were taken at the baseline of KORA S4. BMI was calculated as the individual’s weight in kg divided by the square of their height in metres. Individuals with missing values in the health measures were removed from the dataset.
EpiScore Prediction of Long COVID-19/Hospitalisation
Self-reported COVID-19 phenotypes were available in a subset of individuals from the Generation Scotland DNA methylation sample who had also participated in the CovidLife surveys (N=2,399) [25]. Long COVID-19 phenotypes were ascertained from CovidLife survey 3, (N=1,802 Generation Scotland participants with DNAm profiled), where participants were asked about the total overall time they experienced symptoms in their first/only episode of illness, as well as the whole of their COVID-19 illness. Long COVID-19 was defined here as symptoms persisting for at least 4 weeks from infection and is correct as of February 2021, when the survey 3 was administered. Hospitalisation information, derived from the Scottish Morbidity Records (SMR01), was used to obtain COVID-19 hospital admissions using ICD10 codes U07.1 (lab-confirmed COVID-19 diagnosis), and U07.2 (clinically-diagnosed COVID-19). Hospitalisation data is correct as of February 2021. Logistic regression was used to assess the predictive performance of the T2D EpiScore in relation to long COVID-19 and severe COVID-19 (i.e. hospitalisation), adjusting for sex and age-at-COVID-19 diagnosis. The latter was defined as the age at positive COVID-19 test or 1st January 2021 if COVID-19 test data was not available.
Acknowledgements
This research was funded in whole, or in part, by the Wellcome Trust [104036/Z/14/Z, 108890/Z/15/Z, 216767/Z/19/Z]. For the purpose of open access, the author has applied a CC BY public copyright licence to any Author Accepted Manuscript version arising from this submission.
Generation Scotland received core support from the Chief Scientist Office of the Scottish Government Health Directorates (CZD/16/6) and the Scottish Funding Council (HR03006) and is currently supported by the Wellcome Trust (216767/Z/19/Z). DNA methylation profiling of the Generation Scotland samples was carried out by the Genetics Core Laboratory at the Edinburgh Clinical Research Facility, Edinburgh, Scotland and was funded by the Medical Research Council UK and the Wellcome Trust (Wellcome Trust Strategic Award “STratifying Resilience and Depression Longitudinally” (STRADL; Reference 104036/Z/14/Z). The DNA methylation data assayed for Generation Scotland was partially funded by a 2018 NARSAD Young Investigator Grant from the Brain & Behavior Research Foundation (Ref: 27404; awardee: Dr David M Howard) and by a JMAS SIM fellowship from the Royal College of Physicians of Edinburgh (Awardee: Dr Heather C Whalley). Y.C. is supported by the University of Edinburgh and University of Helsinki joint PhD program in Human Genomics. D.A.G. is supported by funding from the Wellcome Trust 4-year PhD in Translational Neuroscience–training the next generation of basic neuroscientists to embrace clinical research [108890/Z/15/Z]. C.A.V. is a Chancellor’s Fellow funded by the University of Edinburgh. D.L.Mc.C. and R.E.M. are supported by Alzheimer’s Research UK major project grant ARUK-PG2017B−10. R.E.M. is supported by Alzheimer’s Society major project grant AS-PG-19b-010.
Recruitment to the CovidLife study was facilitated by SHARE- the Scottish Health Research Register and Biobank.
SHARE is supported by NHS Research Scotland, the Universities of Scotland and the Chief Scientist Office of the Scottish Government.
The KORA study was initiated and financed by the Helmholtz Zentrum München – German Research Center for Environmental Health, which is funded by the German Federal Ministry of Education and Research (BMBF) and by the State of Bavaria. Furthermore, KORA research has been supported within the Munich Center of Health Sciences (MC-Health), Ludwig-Maximilians-Universität, as part of LMUinnovativ and is supported by the DZHK (German Centre for Cardiovascular Research). The KORA study is funded by the Bavarian State Ministry of Health and Care through the research project DigiMed Bayern (www.digimed-bayern.de).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}