
Abstract
The Coronavirus Disease 2019 (COVID-19) pandemic continues to pose a major public health threat especially in countries with low vaccination rates. To better understand the biological underpinnings of SARS-CoV-2 infection and COVID-19 severity we formed the COVID19 Host Genetics Initiative. Here we present GWAS meta-analysis of up to 125,584 cases and over 2.5 million controls across 60 studies from 25 countries, adding 10 new genome-wide significant loci to the 13 we previously identified1. Genes in novel loci include SFTPD, MUC5B and ACE2, reveal compelling insights regarding disease susceptibility and severity.
Main text
The Coronavirus Disease 2019 (COVID-19) pandemic continues to pose a major public health threat especially in countries with low vaccination rates. To better understand the biological underpinnings of SARS-CoV-2 infection and COVID-19 severity we formed the COVID19 Host Genetics Initiative. Here we present GWAS meta-analysis of up to 125,584 cases and over 2.5 million controls across 60 studies from 25 countries, adding 10 new genome-wide significant loci to the 13 we previously identified1. Genes in novel loci include SFTPD, MUC5B and ACE2, reveal compelling insights regarding disease susceptibility and severity.
Additional genomic regions identified for COVID-19 severity and SARS-CoV-2 infection
Here we present meta-analyses bringing together 60 studies from 25 countries (Figure 1; Supplementary Table 1) for three COVID-19 related phenotypes: (1) individuals critically ill with COVID-19 based on either requiring respiratory support in hospital or who died as a consequence of the disease (9,376 cases - of which 3,197 new in this data release - and 1,776,645 controls), (2) cases with moderate or severe COVID-19 defined as those hospitalized due to symptoms associated with the infection (25,027 cases – 11,386 new - and 2,836,272 controls), and (3) all cases with reported SARS-CoV-2 infection regardless of symptoms (125,584 cases – 76,022 new - and 2,575,347 controls). An overview of the study design is provided in Supplementary Figure 1. We found a total of 23 genome-wide significant loci (P < 5 × 10−8) of which 20 loci remain significant after multiple testing corrections (P < 1.67 × 10−8) to account for the number of phenotypes examined (Figure 2; Supplementary Figure 2; Supplementary Table 2).

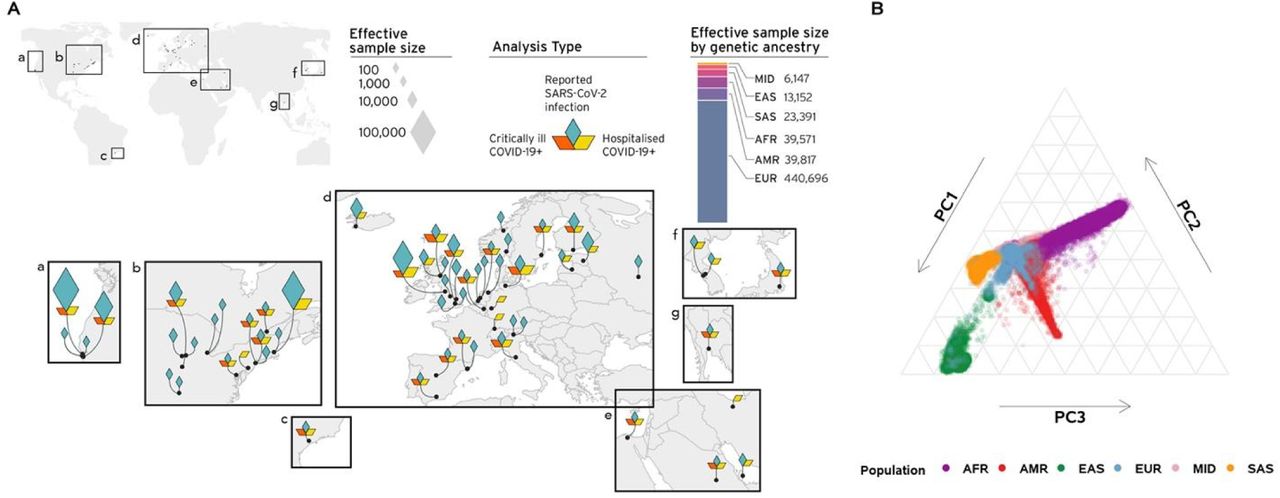
Panel A: Geographical overview of the contributing studies to the COVID-19 HGI and composition by major ancestry groups. Populations are defined as Middle Eastern (MID), South Asian (SAS), East Asian (EAS), African (AFR), Admixed American (AMR), European (EUR). Panel B. Principal components analysis highlights the population structure and the sample ancestry of the individuals participating to the COVID-19 HGI.

A. Top panel shows results of genome-wide association study of hospitalized COVID-19 (n=25,027 cases and n=2,836,272 controls), and bottom panel the results of reported SARS-CoV-2 infection (n=125,584 cases and n=2,575,347 controls). Loci highlighted in yellow (top panel) represent regions associated with severity of COVID-19 manifestation. Loci highlighted in green (bottom panel) are regions associated with susceptibility to SARS-CoV-2 infection. Lead variants for the loci newly identified in this data release are annotated with their respective rsID. B. Results of gene prioritization using different evidence measures of gene annotation. Genes in linkage disequilibrium (LD) region, genes with coding variants and eGenes (fine-mapped cis-eQTL variant PIP > 0.1 in GTEx Lung) are annotated if in LD with a COVID-19 lead variant (r2 > 0.6). V2G: Highest gene prioritized by OpenTargetGenetics’ V2G score. The * indicates SARS-CoV-2 infection susceptibility and + indicates COVID-19 severity. The transparent loci were reported in the previous freeze (data release 5), and loci in bright blue were identified in the current freeze (data release 6).
Heterogeneity in genetic effects across phenotypes, studies, and ancestry groups
Across the genome-wide significant loci, we observed clear patterns of association to the different phenotypes under study. Thus, we developed a two-class Bayesian model for classifying loci based on the patterns of association across the two better-powered phenotypes (COVID-19 hospitalization and SARS-CoV-2 reported infection). Intuitively, loci that are associated to susceptibility will also be associated to severity as to develop COVID-19, SARS-CoV-2 infection needs to first occur. In contrast, those genetic effects that solely modify the course of illness should be associated to severity to illness and not show any association to reported infection except via preferential ascertainment of hospitalized cases in a cohort (Supplementary Methods). We identified 16 loci that are substantially more likely (> 99% posterior probability) to impact the risk of COVID-19 hospitalization and 7 loci that clearly influence susceptibility to SARS-CoV-2 infection (Supplementary Table 3).
We observed that several loci had a significant heterogeneous effect across studies (6/23 loci with P-value for heterogeneity < 2.2×10−3; Supplementary Table 2). Thanks to an increased diversity in our study population (Supplementary Figure 3) we were able to explore if such heterogeneity was due to effect differences across continental ancestry groups. Only one locus (FOXP4) showed a significantly different effect across ancestries (P-value heterogeneity < 7×10−5; Supplementary Table 4 and Supplementary Figure 4), though even at this locus all ancestry groups showed a positive effect estimate. This confirms that factors related to between-study heterogeneity (e.g., variable definition of COVID-19 severity due to different thresholds for testing, hospitalization, and patient recruitment) rather than differences across ancestries are a more likely explanation for the observed heterogeneity in the effect sizes across studies.
Biological insights from novel loci
For the 23 genome-wide significant loci, we explored candidate causal genes and performed a phenome-wide association study to better understand their potential biological mechanisms (Supplementary Table 2,5,6; Supplementary Figure 5). Several of these loci with prior and direct connections to lung disease and SARS-CoV-2 infection mechanism are highlighted here.
Several loci involved in COVID-19 severity implicate lung surfactant biology. A missense variant rs721917:A>G (p.Met31Thr) in SFTPD (10q22.3) confers risk for hospitalization (OR [95% confidence interval [CI] = 1.06 [1.04, 1.08]; P = 1.7 × 10−8) and has been previously associated with increased risk of chronic obstructive pulmonary disease2 (OR = 1.08; P = 2.0 × 10−8), and decreased lung function3 (FEV1/FVC; β = –0.019; P = 2.0 × 10−15). SFTPD encodes the surfactant protein D (SP-D) that participates in innate immune response, protecting the lungs against inhaled microorganisms. The recombinant fragment of SP-D binds to the S1 spike protein of the SARS-CoV-2 and potentially inhibits binding to ACE2 receptor and SARS-CoV-2 infection4. Another missense variant rs117169628:G>A (p.Pro256Leu) in SLC22A31 (16q24.3) also conferring risk for hospitalization (OR [95% CI] = 1.09 [1.06, 1.13]; P = 2.6 × 10−8). SLC22A31 belongs to the family of solute carrier proteins that facilitate transport across membranes5 and is co-regulated with other surfactant proteins6.
We found a variant rs35705950:G>T located in the promoter of MUC5B (11p15.5) to be protective against hospitalization (OR [95% CI] = 0.83 [0.86, 0.93]; P = 6.5 × 10−9). This well-studied promoter variant increases expression of MUC5B in lung in GTEx (P = 6.7 × 10−16) and is the strongest known variant associated with increased risk of developing idiopathic pulmonary fibrosis (IPF)7,8, but also improves survival in IPF patients carrying this mutation9.
Finally, we identified rs190509934:T>C, located 69 bp upstream of ACE2 (Xp22.2) to be associated with decreased susceptibility risk (OR [95% CI] = 0.69 [0.63, 0.75]; P = 3.6 × 10−18). ACE2 is the SARS-CoV-2 receptor and functionally interacts with SLC6A19 and SLC6A20 10, one of which also showed a significant association with susceptibility (rs73062389:G>A at SLC6A20; OR [95% CI] = 1.18 [1.16, 1.20]; P = 2.5 × 10−74). Notably, rs190509934 is 10 times more common in South Asians (MAF = 0.027) than in Europeans (MAF = 0.0024), demonstrating the importance of diversity for variant discovery. Recent results have shown that rs190509934:T>C variant lowers ACE2 expression, which in turn confers protection from SARS-CoV-2 infection11.
We applied Mendelian randomization to infer potential causal relationships between COVID-19 related phenotypes and their genetically correlated traits (Supplementary Methods; Supplementary Table 8,9; Supplementary Figure 6). A novel causal association was observed between genetic liability to type II diabetes (T2D) and SARS-CoV-2 reported infection (OR [95% CI] = 1.02 [1.01, 1.03]; P = 1.6 × 10-3), and COVID-19 hospitalization (OR [95% CI] = 1.06 [1.03, 1.1]; P = 1.4 × 10-4). Multivariable MR (MVMR) was used to estimate the direct effect of liability to T2D on COVID-19-related phenotypes that was not mediated via BMI. This analysis indicated that the observed causal association of liability to T2D on COVID-19 phenotypes is mediated by BMI (Supplementary Table 10).
Discussion
We have substantially expanded the genetic analysis of SARS-CoV-2 infection and COVID-19 severity by doubling the sample size, identifying 10 novel loci. We developed a new approach to systematically assign the 23 discovered loci to either disease susceptibility (7 loci) or disease severity (16 loci). While distinguishing the two phenotypes is challenging because progression to a severe form of the disease requires susceptibility to infection in the first place, it is now evident that the genetic mechanisms involved in these two aspects of the disease can be differentiated. Among the new loci associated with disease susceptibility, ACE2 represents an expected, yet interesting finding. MUC5B, SFTPD, and SLC22A31 are the three most interesting novel loci associated with COVID-19 severity. Their relationship with lung function and lung diseases is consistent with loci previously associated with disease severity. The SPs secreted by alveolar cells, representing an emerging biological mechanism, maintains healthy lung function and facilitates the clearance of pathogens12. The protective effect of the MUC5B variant is unexpected given the otherwise risk-increasing, concordant effect between IPF and COVID-19 observed for other variants8. Nonetheless, this result aligns with the MUC5B promoter variant association that shows a twofold higher survival rate among IPF patients 9. In mice, Muc5b appears essential for effective mucociliary clearance and for controlling infection13 supporting therapies to control mucin secretion to be of potential benefit in COVID-19 patients.
Expanding genomic research to include participants from around the world enabled us to test if the effect of COVID-19 related genetic variants was markedly different across ancestry groups. This was not the case, and we attribute the observed heterogeneity in the effect of COVID-19 related genetic variants to the diverse inclusion criteria across studies in terms of COVID-19 severity.
The novel biological insights gained by this expansion of the COVID-19 Host Genetic Initiative showed that increasing sample size and diversity remain a fruitful activity to better understand the human genetic architecture of COVID-19.
Data Availability
All data produced are available online at https://www.covid19hg.org
Supplementary Methods
Detailed description of methods applied.
Supplementary Figures
Supplementary Figure 1
Analytical summary of the COVID-19 HGI meta-analysis. Using the analytical plan set by the COVID-19 HGI, each individual study runs their analyses and uploads the results to the Initiative, who then runs the meta-analysis. There are three main analyses that each study can contribute summary statistics to: critically ill COVID-19, hospitalized COVID-19 and reported SARS-CoV-2 infection. The phenotypic criteria used to define cases are listed in the dark grey boxes, along with the numbers of cases (N) included in the final all ancestries meta-analysis. Controls were defined in the same way across all three analyses: as everybody that is not a case e.g. population controls (light grey box). Sensitivity analyses, not reported in this Figure, also used mild/asymptomatic COVID-19 cases as controls. Sample number (N) of controls differed between the analyses due to the difference in number of studies contributing data to these.
Supplementary Figure 2
Genome-wide association results for COVID-19 critical illness (Release 6). Results of genome-wide association study of critically ill COVID-19 cases vs population controls (n=9,376 cases and n=1,776,645 controls). Critically ill COVID-19 cases defined as those who required respiratory support in hospital or who were deceased due to the disease.
Supplementary Figure 3
Projection of contributed samples from participating studies into the same PC space. We asked participating studies to perform PC projection using the 1000 Genomes Project and Human Genome Diversity Project as a reference, with a common set of variants. For each panel (except for the reference), colored points correspond to contributed samples from each cohort, whereas gray points correspond to the 1000 Genomes reference samples. Color represents a genetic population that each cohort specified. Since 23andme, genomicsengland100kgp, and MVP only submitted PCA images, we overlaid their submitted transparent images using the same coordinates, instead of directly plotting them.
Supplementary Figure 4
Forest plots for highly heterogeneous loci. Forest plots for 6/23 loci that showed a significant heterogeneous effect across studies. For each of the loci, effect sizes and confidence intervals are reported for each contributing study. Studies are grouped by ancestry, with summary effects reported for each ancestry subgroup. The summary effect size across all studies is also reported in the bottom panel for each locus.
Supplementary Figure 5
LocusZoom plots to visualize the meta-analysis results at the loci passing genome-wide significance. For each genome-wide significant locus in three meta-analyses: meta-analysis of critical illness, hospitalization, and reported infection, we showed 1) a Manhattan plot of each locus where a color represents a weighted-average r2 value (see COVID-19 Host Genetics Initiative, 2021) to a lead variant (unadjusted P-values from the two-tailed inverse variance weighted meta-analysis); 2) r2 values to a lead variant across gnomAD v2 populations, i.e., African/African-American (AFR), Latino/Admixed American (AMR), Ashkenazi Jewish (ASJ), East Asian (EAS), Estonian (EST), Finnish (FIN), Non-Finish Europeans (NFE), North-Western Europeans (NWE), and Southern Europeans (SEU); 3) genes at a locus; and 4) genes prioritized by each gene prioritization metric where a size of circles represents a rank in each metric. Note that the COVID-19 lead variants were chosen across all the meta-analyses (Supplementary Table 2) and were not necessarily a variant with the most significant P-value from each inverse variance weighted meta-analysis.
Supplementary Figure 6
Genetic correlations and Mendelian randomization causal estimates between 38 traits and COVID-19 critical illness, hospitalization, and SARS-CoV-2 reported infection. Larger squares correspond to more significant P-values, with genetic correlations or MR causal estimates significantly different from zero at a P < 0.05 shown as a full-sized square. Genetic correlations or causal estimates that are significantly different from zero at a false discovery rate (FDR) of 5% are marked with an asterisk. Two-sided P-values were calculated using LDSC for genetic correlations and Inverse variance weighted analysis for MR.
Supplementary Tables
Supplementary Table 1. Information regarding studies contributing to the consortium. All independent studies that contributed data to at least one of the three main all-ancestries meta-analyses are listed, with more detailed information where available. The COVID-19 HGI analysis plan can be found on the consortium’s website https://www.covid19hg.org.
Supplementary Table 2. Genome-wide significant results from three COVID-19 phenotype meta-analyses. Loci that reached genome-wide significance (P < 1.67 × 10−8) in at least one of our three inverse-variance weighted meta-analyses of COVID-19 phenotypes (results for each loci listed for all three phenotypes): hospitalization due to COVID-19 (n=25,027 cases and n=2,836,272 controls), and reported SARS-COV-2 infection (n=125,584 cases and n=2,575,347 controls). Associations that did not reach significance threshold of P < 1.67 × 10-8 in the particular analysis are colored in grey. Effect allele frequency is the sample size weighted frequency across studies included in each meta-analysis. P-value is reported for meta-analysis variant association with trait (P-value association). Suggested phenotypic impact of each locus is probabilistically assigned as “Disease severity” or “Infection susceptibility”, as described in the Supplementary Methods and Supplementary Table 3. Between-study heterogeneity P-values (from a two-tailed Cochran’s Q test) are reported only for the analysis corresponding to the suggested phenotype. Genes in linkage disequilibrium (LD) region: Genes that overlap with a genomic range that contains any variants in LD (r2 > 0.6) with each lead variant. A gene with a minimum distance to gene body is bolded. Genes with coding variants: Genes with a loss-of-function or missense variant in LD with a lead variant (r2 > 0.6). eGenes: Genes with a fine-mapped cis-eQTL variant (PIP > 0.1) in GTEx and eQTL catalog that is in LD with a lead variant (r2 > 0.6). V2G: Highest gene prioritized by OpenTargetGenetics’ V2G score.
Supplementary Table 3. Probabilistic assignment of variants into disease severity vs infection susceptibility. We report the results of the phenotypic impact assignment for each of the genome-wide significant variants reported in Supplementary Table 2. Beta, standard error and p-values are reported for a restricted meta-analysis of hospitalized COVID19 cases (n cases = 23,988 cases, n controls = 2,834,885) and reported SARS-Cov-2 infection (n=114,516 cases, n=2,138,237 controls), including only studies that only contributed to both analysis. Posterior probabilities are reported, to belong to either a disease severity (prob_SEV) or infection susceptibility model (prob_SEV). The phenotypic impact of each variant is assigned with a probability cut-ff of 99%.
Supplementary Table 4. Between-ancestry heterogeneity test for COVID-19 meta-analysis lead variants. We report lead variant summary information for each locus reported in Supplementary Table 2, with meta-analysis results stratified by genetic ancestry (AFR=African, AMR=Ad-mixed American, EAS= East Asian, EUR=European, MID=Middle Eastern, SAS=South Asian). Meta-analysis P-values reported are unadjusted values from a two-tailed inverse-variance weighted meta-analysis. Between-analysis heterogeneity P-values from a chi-squared test of the Cochran’s Q measure of the effect sizes. Effect allele frequency is the sample size weighted frequency across studies included in each meta-analysis. Sample sizes and number of studies for each ancestry are reported in the table.
Supplementary Table 5. eGenes in GTEx v8 or eQTL catalogue. Genes with at least one fine-mapped cis-eQTL variant (PIP > 0.1) that is in LD with a lead variant (r2 > 0.6) in any tissue of GTEx v8 or eQTL catalogue. The r2 column represents the r2 value between the COVID-19 lead variant and eQTL variant.
Supplementary Table 6. PheWAS associations for GWAS lead variants in LD (r2>0.6) with COVID-19 index variants. We report phenotype associations for variants which are lead variants of their respective phenotype and in LD (r2>0.6) with COVID-19 associated SNPs. The associations were retrieved from OpenTargetsGenetics, UK Biobank and FinnGen.
Supplementary Table 7. SNP heritability estimated for three meta-analysis phenotypes. We used LD score regression (LDSC) to estimate SNP heritability from the European-only summary statistics and All ancestries summary statistics, for our three meta-analysis phenotypes. Heritability P-value was calculated from a two-sided z-distribution. All analyses were performed using European panel LD scores.
Supplementary Table 8. Genetic correlation results between complex traits and COVID phenotypes (EUR only). Two-sided P-values were obtained using LD score regression and adjustments for multiple comparisons were made using FDR. Phenotypes surviving FDR are bolded.
Supplementary Table 9. Causal association of complex traits on COVID-19 phenotypes (EUR only). Two-sided P-values for the primary analysis were obtained using a fixed-effects inverse weighted analysis, with multiple comparisons adjusted for using FDR. Two-sided P-values for the sensitivity analyses were obtained using weighted median estimator (WME), weighted mode based estimator (WMBE), MR Egger regression and MR-PRESSO outlier corrected estimates.
Supplementary table 10 A. Direct effects of BMI and type 2 diabetes on risk of SARS-CoV-2 phenotypes using Multivariable Mendelian Randomization. 10B. Test for weak instruments using Sanderson-Windmeijer conditional F-statistics in multivariable MR.
Supplementary Table 11. Study specific acknowledgements and competing interests.




{kind=link}
{kind=link}