
Abstract
SARS-CoV-2 variants of interest have been associated with increased transmissibility, neutralization resistance and disease severity. Ongoing SARS-CoV-2 genomic surveillance world-wide has improved our ability to rapidly identify such variants. Here we report the identification of a potential variant of interest assigned to the PANGO lineage C.1.2. This lineage was first identified in May 2021 and evolved from C.1, one of the lineages that dominated the first wave of SARS-CoV-2 infections in South Africa and was last detected in January 2021. C.1.2 has since been detected across the majority of the provinces in South Africa and in seven other countries spanning Africa, Europe, Asia and Oceania. The emergence of C.1.2 was associated with an increased substitution rate, as was previously observed with the emergence of the Alpha, Beta and Gamma variants of concern (VOCs). C.1.2 contains multiple substitutions (R190S, D215G, E484K, N501Y, H655Y and T859N) and deletions (Y144del, L242-A243del) within the spike protein, which have been observed in other VOCs and are associated with increased transmissibility and reduced neutralization sensitivity. Of greater concern is the accumulation of additional mutations (C136F, Y449H and N679K) which are also likely to impact neutralization sensitivity or furin cleavage and therefore replicative fitness. While the phenotypic characteristics and epidemiology of C.1.2 are being defined, it is important to highlight this lineage given its concerning constellations of mutations.
Main Text
More than a year into the COVID-19 pandemic, SARS-CoV-2 remains a global public health concern. Ongoing waves of infection result in the selection of SARS-CoV-2 variants with novel constellations of mutations within the viral genome1–4. Some emerging variants accumulate mutations within the spike region that result in increased transmissibility and/or immune evasion, making them of increased public health importance2–4. Depending on their clinical and epidemiological profiles, these are either designated as variants of interest (VOI) or variants of concern (VOC)5, and ongoing genomic surveillance is essential for early detection of such variants. There are currently four VOCs (Alpha, Beta, Gamma and Delta) and four VOIs (Eta, Iota, Kappa and Lambda) in circulation globally. Of these, Alpha, Beta and Delta have had the most impact globally in terms of transmission and immune evasion, with Delta rapidly displacing other variants to predominate globally, including in South Africa.
Ongoing genomic surveillance in South Africa also detected an increase in sequences assigned to C.1 during the third wave of SARS-CoV-2 infections in May 2021, which was unexpected since C.1, first identified in South Africa6,7, was last detected in January 2021. Upon comparison of the mutational profiles between these and older C.1 sequences (which only contain the D614G mutation within the spike), it was clear that these new sequences had mutated substantially. C.1 had minimal spread globally but was detected in Mozambique and had accumulated additional mutations resulting in the PANGO lineage C.1.17. These new sequences, however, were also very distinct from C.1.1, resulting in the assignment of the PANGO lineage C.1.2 on 22 July 20218. C.1.2 is highly mutated beyond C.1 and all other VOCs and VOIs globally with between 44-59 mutations away from the original Wuhan Hu-1 virus (Fig. 1a). While the VOI Lambda (C.37) is phylogenetically closest to C.1.2, the latter has distinct lineage-defining mutations.

a, Phylogenetic tree of 5,756 global sequences, including Variants of Concern (VOC), Variants of Interest (VOI), and the C.1.2 lineage, colored according to the key. Of these, 1,922 sequences are from South Africa. The tree is scaled by divergence (number of mutations) and colored by Nextstrain clade (shown in the key). The C.1.2 lineage (purple) forms a distinct, highly mutated cluster within clade 20D. b, Magnified time-scaled phylogenetic sub-tree of C.1.2 sequences with >=95% coverage data (n=54) detected across the globe, colored by province (circles for South African sequences) or by country (squares for non-South African sequences). c, Clock estimate of lineage evolution during the SARS-CoV-2 pandemic. C.1.2 samples are indicated by purple dots; all other samples are indicated by branches only. The regression line represents the average mutation rate of the SARS-CoV-2 sequences in the tree (26.6 substitutions/year). C.1.2 sequences form a sub-cluster above the regression line, suggesting an increased substitution rate above the average.
The C.1.2 lineage was first detected in the Mpumalanga and Gauteng provinces of South Africa, in May 2021 (Fig. 1b and Supplementary Fig. 1a). In June 2021, it was also detected in the KwaZulu-Natal and Limpopo provinces of South Africa as well as in England and China (Fig. 1b and Supplementary Fig. 1b). As of August 13, 2021 the C.1.2 lineage has been detected in 6/9 South African provinces (including the Eastern Cape and Western Cape), the Democratic Republic of the Congo (DRC), Mauritius, New Zealand, Portugal and Switzerland (Fig. 1b and Supplementary Fig. 1b and c).
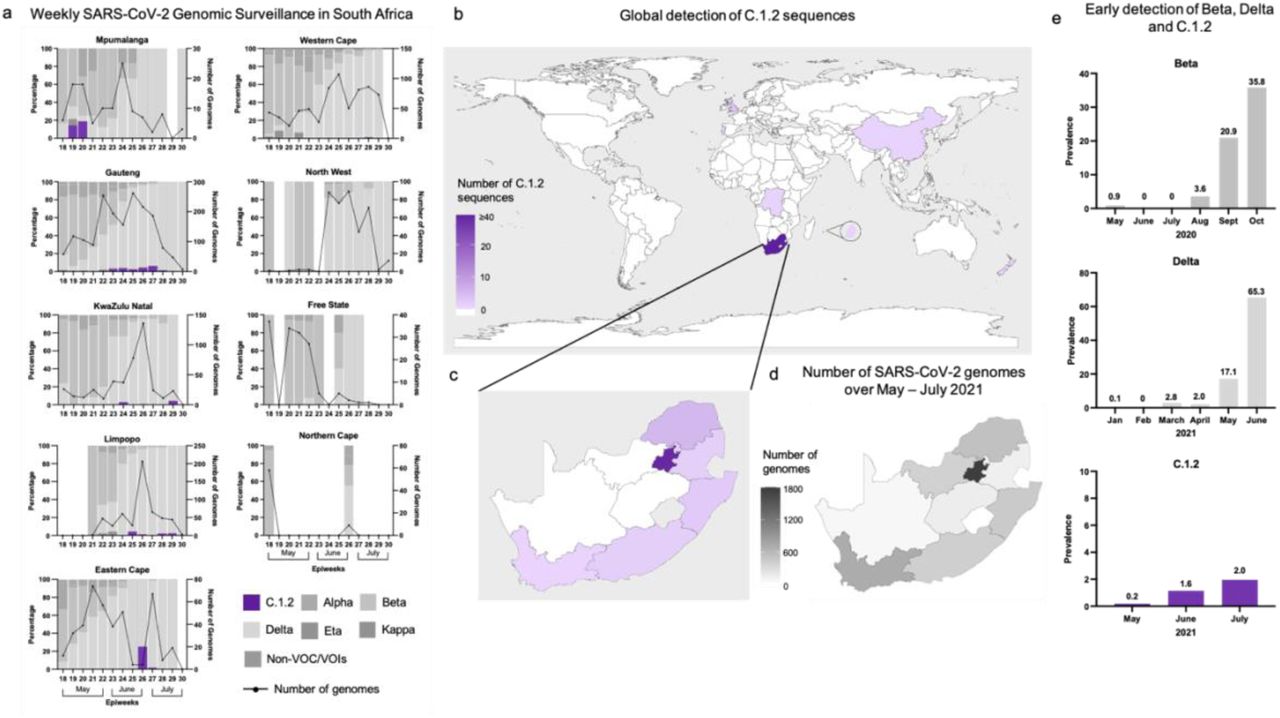
Maps showing the locations in which C.1.2 sequences have been detected, colored according to the number of C.1.2 sequences identified/sequenced. a, Percentage of genomes that are assigned to various SARS-CoV-2 lineages in South Africa for each of the provinces, with C.1.2 shown in purple, by epidemiological week (epiweek) for the months of May - July 2021. The number of genomes sequenced for each epiweek is shown by the black line. b, Global map highlighting South Africa, England, Portugal, Switzerland, China, the Democratic Republic of the Congo, Mauritius (shown in the magnified bubble) and New Zealand, across which 63 C.1.2 sequences have been detected. c, Map of South Africa highlighting the provinces in which C.1.2 has been detected, colored using the same color key as panel a. d, Map of South Africa showing the number of SARS-CoV-2 genomes (n=4,953 as of 13 August 2021) that have been sequenced by province in the months of May, June and July 2021, during which C.1.2 has been detected. e, Early prevalence rates of Beta, Delta and C.1.2 in South Africa based on the number of SARS-CoV-2 sequences generated for each month.
As of August 13, 2021 we have identified 63 sequences that match the C.1.2 lineage, of which 59 had sufficient sequence coverage to be used in phylogenetic analyses and/or spike analysis. All C.1.2 sequences including those with poor coverage (from the DRC and Mpumalanga) can be found on GISAID (www.gisaid.org), the global reference database for SARS-CoV-2 viral genomes9,10, and listed in Supplementary Tables 1 and 2. The majority of these sequences (n=53) are from South Africa. Though SARS-CoV-2 genomic surveillance is ongoing, there is normally a delay of 2-4 weeks between sampling and data being publicly available on GISAID. Provincial detection of C.1.2 to some extent mirrored the depth of sequencing across SA (Supplementary Fig. 1a, c and d), suggesting that it may be present in under-sampled provinces and these numbers are most likely an underrepresentation of the spread and frequency of this variant within South Africa and globally. Nevertheless, we see consistent increases in the number of C.1.2 genomes in South Africa on a monthly basis, where in May C.1.2 accounted for 0.2% (2/1054) of genomes sequenced, in June 1.6% (25/2177) and in July 2.0% (26/1326), similar to the increases seen in Beta and Delta in South Africa during early detection (Supplementary Fig. 1e).
Provided are the GISAID strain name and gisaid_epi_isl accession numbers for sequences with good quality used in the phylogenetic trees and highlighter plots and potential C.1.2 sequences that have not been used due to poor sequence coverage (shown as None*). All sequences below were used in local distribution plots (Supplementary Fig. 1).
We gratefully acknowledge the following authors from the originating laboratories responsible for obtaining the specimen, as well as the submitting laboratories where the genomes were generated and shared via GISAID, on which this research is based. All submitters of data may be contacted via www.gisaid.org. Authors are listed according to how they were provided on GISAID. Listed are those with good quality used in the phylogenetic trees and highlighter plots and potential C.1.2 sequences that have not been used due to poor sequence coverage (shown as None*). All sequences below were used in global distribution plots (Supplementary Fig. 1).
Preliminary molecular clock estimates suggested that the overall rate of evolution of SARS-CoV-2 in 2020 was 8×10−4 substitutions/site/year, which equates to 24 substitutions per year11. The current global estimate (derived from a global Nextstrain build, https://nextstrain.org/ncov/gisaid/global, accessed August 15th 2021) including multiple variants of concern/interest suggests a similar rate of approximately 25.2 substitutions per year (8.4×10−4 substitutions/site/year). The global phylogeny, including C.1.2 sequences, gives a slightly higher clock rate of 26.6 substitutions per year (8.9×10−4 substitutions/site/year), with the C.1.2 sequences clearly having a higher substitution rate than the majority of other sequences (Fig. 1c). To obtain an estimate of the rate of C.1.2 specifically, we performed a root-to-tip regression of C.1.2 against C.1 sequences. This suggested that the emergence of the C.1.2 lineage resulted from a rate closer to 1.4×10−3, or ∼41.8 mutations per year, which is approximately 1.7-fold faster than the current global rate and 1.8-fold faster than the initial estimate of SARS-CoV-2 evolution. This short period of increased evolution compared to the overall viral evolutionary rate was also associated with the emergence of the Alpha, Beta and Gamma VOCs2,3,12, suggesting a single event, followed by the amplification of cases, which drove faster viral evolution13.
C.1.2 shares some mutations with C.1 but has accumulated additional mutations within the ORF1ab, spike, ORF3a, ORF9b, E, M and N proteins (Fig. 2a). Of these mutations, 30 occur in >50% of the sequences. Several mutations were observed within the spike protein, with >50% of the viruses assigned to C.1.2 having 14 mutations, including five within the NTD (C136F, Y144del, R190S, D215G and 242-243del (L242 and A243 deletions)), three within the receptor binding motif (RBM) (Y449H, E484K and N501Y) and two adjacent to the furin cleavage site (N679K and T716I). P9L, D614G, H655Y and T859N make up the remaining four major mutations. Though these mutations occur in the majority of C.1.2 viruses, there is additional variation within the spike region of this lineage (Fig. 2b), suggesting ongoing intra-lineage evolution. Approximately 44% of the viruses also contain a P25L mutation in the NTD, ∼19% have L585F in S1, ∼16% have T478K in the RBM, ∼11% contain P681H adjacent to the furin cleavage site, 8% have D936H, and a further ∼8% have H1101Q in S2. The majority of these mutations (P9L, C136F, R190S, D215G, L242del, A243del, Y449H, E484K, N501Y, H655Y, and T716I) appeared together early in the lineage evolution (Fig. 3a). Thereafter, the majority of sequences have also accumulated the mutations Y144del, N679K and T859N. The mutations P25L, W152R, R346K, T478K, L585F, N440K, P681H, A879T, D936H and H1101Q can be seen in some of the smaller clusters from more recent sequences, further highlighting continued evolution within the lineage.

a, Full genome representation of C.1.2 showing all mutations, with those in the spike (green) colored according to functional regions, including N-terminal domain (NTD, blue), receptor binding domain (RBD, red), receptor binding motif (RBM, purple), subdomain 1 and 2 (SD1 or SD2, grey) and the cleavage site (S1/S2, yellow). Figure generated by covdb.stanford.edu. b, Highlighter plot of C.1.2 spike sequences with >=90% coverage of the spike region (n=57) identified across the globe, labelled according to the location identified and sequence name. Representative sequence shown in a, is labelled in bold. Mismatches compared to the C.1 strain are colored by Se-Al in hiv.lanl.gov highlighter tool as shown in the key. Large dark grey regions represent missing sequence data. Regions within the spike are colored as in panel a.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
a, A phylogenetic tree highlighting the introduction of spike mutations in the different sub-clades of the C.1.2 lineage. The tree is annotated with date of collection and colored according to location (country or South African province as indicated in the key) (Figure generated from a Nextstrain Build of global C.1.2 sequences with >=95% coverage data). b, Visualization of C.1.2 lineage-defining mutations shared with Variants of Concern (VOC) and Variants of Interest (VOI). All C.1.2 mutations are shown, with those present in >50% of C.1.2 sequences in dark purple and those present in <50% of C.1.2 sequences in light purple. For VOCs and VOIs only mutations present in at least 50% of sequences are shown (as determined by frequency information at outbreak.info). VOC and VOI mutations are colored by the Nextstrain clade. c, Schematic showing C.1.2 mutations on the RBD-down conformation of SARS-CoV-2 spike, with domains of a single protomer shown in cartoon view and colored cyan (N-terminal domain, NTD), red (C-terminal domain/receptor binding domain, CTD/RBD), grey (subdomain 1 and 2, SD1 and SD2), and green (S2). The adjacent protomers are shown in translucent surface view and colored shades of grey. Lineage-defining mutations (found in >50% of sequences) are colored dark purple, with additional mutations (present in <50% of sequences) colored light purple. Key mutations known/predicted to influence neutralization sensitivity (C136F and P25L, Y144del, L242del/A243del, and E484K), or furin cleavage (H655Y and N679K) are indicated. Image was created using the PyMOL molecular graphic program.
Several (52%, 13/25) of the spike mutations identified in C.1.2 have previously been identified in other VOIs and VOCs (Fig. 3b). These include D614G, common to all variants14, and E484K and N501Y which are shared with Beta and Gamma, with E484K also seen in Eta and N501Y in Alpha. The T478K substitution is seen in <50% of the C.1.2 viruses but is also observed in Delta. N440K and Y449H co-localize on the same outer face of C.1.2 RBD (Fig. 3c). While these mutations are not characteristic of current VOCs/VOIs, they have been associated with escape from certain class 3 neutralizing antibodies15,16. The combination of these mutations presents a potentially novel antigenic landscape for C.1.2 variant specific antibodies. More striking, however, was the remodeling of NTD relative to the Wuhan Hu-1 sequence (blue, Fig. 3c). While Y144del and 242-244del cause frameshifts to the immunodominant N3 or N5 loops of NTD in the Alpha or Beta variants respectively17, the deletion of both regions in C.1.2 (with a different N5 frameshift relative to Beta) likely contributes to evading NTD immune responses elicited by infection with either Alpha or Beta. Furthermore, the C136F mutation abolishes a disulphide bond with the N1 loop of NTD, and in combination with P25L likely contributes to immune escape by conformationally liberating the entire N-terminus of NTD. Mutations close to the furin cleavage site have also been observed in VOCs, H655Y has been seen in Gamma and P681R/H have been seen in Alpha, Delta, and Kappa (S1/S2 region in Fig. 3c). In the C.1.2 lineage, N679K and P681H are mutually exclusive (with N679K predominating) and may therefore perform a similar role by increasing the local, relative positive charge and improving furin cleavage. Evolution involving the introduction of N679K or P681H has recently been seen within Gamma (P.1)18. The identification of convergent evolution between C.1.2 and other VOIs and VOCs suggests that this variant may also share concerning phenotypic properties with VOCs.
We are currently assessing the impact of this variant on antibody neutralization following SARS-CoV-2 infection or vaccination against SARS-CoV-2 in South Africa.
Discussion/Conclusion
We have identified a new SARS-CoV-2 variant assigned to the PANGO lineage C.1.2. This variant has been detected throughout the third wave of infections in South Africa from May 2021 onwards and has been detected in seven other countries within Europe, Asia, Africa and Oceania. The identification of novel SARS-CoV-2 variants is commonly associated with new waves of infection. Like several other VOCs, C.1.2 has accumulated a number of substitutions beyond what would be expected from the background SARS-CoV-2 evolutionary rate. This suggests the likelihood that these mutations arose during a period of accelerated evolution in a single individual with prolonged viral infection through virus-host co-evolution19–21. Deletions within the NTD (like Y144del, seen in C.1.2 and other VOCs) have been evident in cases of prolonged infection, further supporting this hypothesis22–24.
C.1.2 contains many mutations that have been identified in all four VOCs (Alpha, Beta, Delta and Gamma) and three VOIs (Kappa, Eta and Lambda) as well as additional mutations within the NTD (C136F), RBD (Y449H), and adjacent to the furin cleavage site (N679K). Many of the shared mutations have been associated with improved ACE2 binding (N501Y)25–29 or furin cleavage (H655Y and P681H/R)30–32, and reduced neutralization activity (particularly Y144del, 242-244del, and E484K)17,33–39, providing sufficient cause for concern of continued transmission of this variant. Future work aims to determine the functional impact of these mutations, which likely include neutralizing antibody escape, and to investigate whether their combination confers a replicative fitness advantage over the Delta variant.
The C.1.2 lineage is continuing to grow. At the time of submission (20 August 2021) there were 80 C.1.2 sequences in GISAID with it now having been detected in Botswana and in the Northern Cape of South Africa.
Methods
Sampling of SARS-CoV-2 and Metadata
As part of monitoring the viral evolution by the Network for Genomics Surveillance of South Africa (NGS-SA)40, seven sequencing hubs receive randomly selected samples for sequencing every week according to approved protocols at each site. These samples include remnant nucleic acid extracts or remnant nasopharyngeal and oropharyngeal swab samples from routine diagnostic SARS-CoV-2 PCR testing, from public and private laboratories in South Africa. Permission was obtained for associated metadata for the samples including date and location (district and province) of sampling, and sex and age of the patients to offer additional insights about the epidemiology of the infection caused by the virus.
Ethical statement
The project was approved by the University of the Witwatersrand Human Research Ethics Committee (HREC) (ref. M180832, M210159, M210752), University of KwaZulu–Natal Biomedical Research Ethics Committee (ref. BREC/00001510/2020), Stellenbosch University HREC (ref. N20/04/008_COVID19) and the University of Cape Town HREC (ref. 383/2020) and the University of Pretoria, Faculty of Health human ethics committee, (ref H101-2017). Individual participant consent was not required for the genomic surveillance. This requirement was waived by the Research Ethics Committees.
Whole-genome sequencing and genome assembly
RNA Extraction
RNA was extracted either manually or automatically in batches, using the QIAamp viral RNA mini kit (QIAGEN, California, USA) as per manufacturer’s instructions or the Chemagic 360 using the CMG-1049 kit (PerkinElmer, Massachusetts, USA), respectively. A modification was done on the manual extractions by adding 280 µl per sample, in order to increase yields. 300 µl of each sample was used for automated magnetic bead-based extraction using the Chemagic 360. RNA was eluted in 60 µl of the elution buffer. Isolated RNA was stored at - 80°C prior to use.
PCR and library preparation
Sequencing was performed using the COVIDSeq or nCoV-2019 ARTIC network sequencing protocol (https://artic.network/ncov-2019), which is an amplicon-based next-generation sequencing approach (Illumina, Inc, USA)41. Briefly, the first strand synthesis was carried out on extracted RNA samples using random hexamers primers from the SuperScript IV reverse transcriptase synthesis kit (Life Technologies). The synthesized cDNA was amplified using two separate multiplex polymerase chain reactions (PCRs), producing 98 amplicons across the SARS-CoV-2 genome. The primer pool additionally had primers targeting human RNA, producing an additional 11 amplicons. The pooled PCR products underwent bead-based tagmentation where they get fragmented and tagged to the adapter sequences using the Nextera Flex DNA library preparation kit. The adapter-tagged amplicons were cleaned-up using AmpureXP purification beads (Beckman Coulter, High Wycombe, U and amplified using one round of PCR. The PCRs were indexed using the Nextera CD indexes (Illumina, Sand Diego, CA, USA) according the manufacturer’s instructions. Tagged libraries were pooled and cleaned using the K). Pooled samples were quantified using Qubit 3.0 or 4.0 fluorometer (Invitrogen Inc.) using the Qubit dsDNA High Sensitivity assay according to manufacturer’s instructions. The fragment sizes were analyzed using TapeStation 4200 (Invitrogen). The pooled libraries were further normalized to 4nM concentration and 25 μl of each normalized pool containing index adapter sets 1, 2, 3, and 4 were combined in a new tube. The final library pool was denatured and neutralized with 0.2N sodium hydroxide and 200 mM Tris-HCL (pH7), respectively. 1.5 pM sample library was spiked with 2% PhiX. Libraries were loaded onto a 300-cycle NextSeq 500/550 HighOutput Kit v2 and run on the Illumina NextSeq 550 instrument (Illumina, San Diego, CA, USA).
Assembly, processing and quality control of genomic sequences
Raw reads from Illumina sequencing were assembled using the Exatype NGS SARS-CoV-2 pipeline v1.6.1, (https://sars-cov-2.exatype.com/) or Genome Detective 1.132/1.133 (https://www.genomedetective.com/) and the Coronavirus Typing Tool42,43. Samples sequenced from Oxford Nanopore GridION were assembled according to the Artic-nCoV2019 novel coronavirus bioinformatics protocol (https://artic.network/ncov-2019/ncov-2019-bioinformatics-sop.html). For these samples raw reads were base called and demultiplexed using Guppy. To guarantee accuracy of the base calls, we only used dual indexed reads (i.e. required barcodes both ends). A reference-based assembly and mapping was generated for each sample using Minimap2 and consensus calculated using Nanopolish. The reference genome used throughout the assembly process was NC_045512.2 (Accession number: MN908947.3). The initial assembly obtained was cleaned by aligning mapped reads to the references and filtering out low-quality mutations using the Geneious software v2021.0.3 (Biomatters). Quality control reports were obtained from Nextclade44. The resulting consensus sequence was further manually polished by considering and correcting indels in homopolymer regions that break the open reading frame (probably sequencing errors) using Aliview v1.27, (http://ormbunkar.se/aliview/)45. Mutations resulting in mid-gene stop codons and frameshifts were reverted to wild type. Regions with clustered mutations and deletions resulting in frameshifts where annotated as gaps and insertions where removed. Sequences with less than 80% coverage relative to the Wuhan-Hu-1 reference where discarded. All assemblies were deposited in GISAID (https://www.gisaid.org/)10 and the GISAID accession was included as part of Supplementary Table 1. Clade and lineage assignment was determined using Nextclade and Pangolin46.
Classification of lineage, clade and associated mutations
The ‘Phylogenetic Assignment of Named Global Outbreak Lineages’ (PANGOLIN) software suite (https://github.com/hCoV-2019/pangolin) was used for the dynamic SARS-CoV-2 lineage classification46. The SARS-CoV-2 genomes in our dataset were also classified using the clade classification proposed by NextStrain (https://nextstrain.org/) built for real-time tracking of the pathogen evolution47. The PANGO lineage identified predominantly in South Africa in this study is now assigned to the lineage C.1.2 (Pangolin version v3.1.7, lineages version 2021-07-28); the corresponding Nextclade classification is 20D (Nextclade version v1.5.3, clades version 2021-07-28). The C.1.2 lineage and its associated mutations were further confirmed using the Stanford Coronavirus Antiviral & Resistance Database (CoVDB) (https://covdb.stanford.edu/) and Outbreak.info (https://outbreak.info/).
Dataset Compilation
At the time of writing, there were over 2.9 million SARS-CoV-2 genomes available on GISAID (https://www.gisaid.org). Due to the size of this dataset, sub-sampling was performed to obtain a representative but manageable sample of genomes. A preliminary dataset was downloaded from GISAID; the options ‘complete’, ‘high coverage’, and ‘collection date complete’ were selected to ensure that only genomes with complete date information and less than 5% N content were included. This contained all C.1.2 genomes, genomes from the C.1 lineage (the original lineage to which C.1.2 was assigned), the C.1.1 lineage (a Mozambican lineage that evolved from C.17, and South African. The global and African Auspice datasets were also downloaded (accessed 13 August 2021). This dataset was further down-sampled using a custom build of the Nextstrain SARS-CoV-2 pipeline47 to produce a final dataset of 5,756 genomes. Of these, 54 are from lineage C.1.2. Due to the fact that C.1.2 was first detected and is most prevalent in South Africa, we chose to include a large proportion of South African sequences, resulting in 1,922 South African genomes. To include global context, there were an additional 946 sequences from the rest of Africa, 843 from Asia, 1,038 from Europe, 443 from South America, 376 from North America, and 188 from Oceania. This dataset included genomes from all Variants of Concern (VOC) and Variants of Interest (VOI) as defined by the WHO5.
Temporal Analysis
We conducted temporal analysis to ensure that C.1.2 possesses a strong enough temporal signal for dated phylogenetic analysis, as well as to get an estimate of the molecular clock rate for the C.1.2 lineage. To do this, 54 C.1.2 and 135 C.1 samples were extracted from the initial dataset and aligned within MAFFT48. The alignment was manually inspected in AliView45 to ensure there were no errors. IQ-TREE49 was used to construct an undated maximum likelihood phylogeny of C.1 and C.1.2 samples, using the HKY+I nucleotide substitution model. The resulting tree was analyzed in TempEst50 for the presence of a temporal signal. Inspection of the tree revealed a small cluster of sequences from several countries that formed a monophyletic group distinct from other C.1 and C.1.2 samples. Further inspection of these sequences with CoVDB (https://covdb.standford.edu/) showed that they contain several spike mutations not characteristic of either C.1 or C.1.2, suggesting they have been mis-assigned. There were also several samples that may violate the molecular clock assumption. These sequences were removed and the tree remade. The final tree showed a strong positive temporal signal, with a correlation coefficient of 0.97 and R2 of 0.95. The slope of the regression suggested a preliminary clock rate estimate of 1.4×10−3.
Phylogenetic Analysis
Phylogenetic analysis was conducted with a custom Nextstrain SARS-CoV-2 build47. Briefly, the pipeline filters sequences, aligns these sequences with Nextalign (https://github.com/neherlab/nextalign), sub-samples the datasets (resulting in the dataset described above), constructs a phylogenetic tree with IQ-TREE49, refines and dates the tree with TreeTime51, reconstructs ancestral states, and assigns Nextstrain clades to the sequences. The tree was visualized with Auspice to confirm the presence of a C.1.2 cluster. This revealed that several non-C.1.2 samples clustered with C.1.2. These sequences were inspected for the presence of the major C.1.2 mutations (dark purple mutations in Fig. 3b). All sequences possessed at least eight major mutations; this, along with the clustering, was used as evidence to re-assign the sequences to C.1.2, resulting in a set of 54 C.1.2 genomes.
SARS-CoV-2 Model
We modelled the spike protein on the basis of the Protein Data Bank coordinate set 7A94. We used the Pymol program (The PyMOL Molecular Graphics System, version 2.2.0) for visualization.
Data availability
All of the global SARS-CoV-2 genomes of the C.1.2 lineage generated and presented in this article are publicly accessible through the GISAID platform (https://www.gisaid.org/), along with all other SARS-CoV-2 genomes generated by the NGS-SA. The GISAID accession identifiers of the C.1.2 sequences analyzed in this study are provided as part of Supplementary Tables 2 and 3, which also contain the metadata for the sequences. The nextstrain build of C.1.2 and global sequences will be made available at https://nextstrain.org/groups/ngs-sa.
Acknowledgements
We acknowledge additional NGS-SA members: Adriano Mendes, Allison Glass, Amy Strydom, Arash Iranzadeh, Bulelani Manene, Cheryl Cohen, Deelan Doolabh, Derek Tshiabuila, Diana Hardie, Dominique Goadhals, Gert van Zyl, Innocent Madau, Kamela Mahlakwane, Kathleen Subramoney, Kruger Marais, Linda de Gouveia, Lynn Tyers, Michaela Davids, Noluthando Duma, Rageema Joseph, Yajna Ramphal, Upasana Ramphal, Sibongile Walaza, Simnikiwe Mayaphi, Stephen Korsman, Susan Engelbrecht, Tania Stander, Terry Marshall, Zinhle Makatini. We thank our colleagues at both private and public testing laboratories, who submit samples for sequencing despite numerous challenges. We would like to acknowledge the teams within the National Institute for Communicable Diseases Centre for Respiratory Diseases and Meningitis, Sequencing Core Facility and Centre for HIV and STIs. In addition, we thank Hyrax Biosciences for the use of their Exatype platform, Bridge-the-Gap and the Cape Town Immunology Laboratory. The Network for Genomic Surveillance South Africa (NGS-SA) is supported by the Strategic Health Innovation Partnerships Unit of the South African Medical Research Council, with funds received from the South African Department of Science and Innovation. Sequencing activities for the different sequencing hubs were provided by a conditional grant from the South African National Department of Health as part of the emergency COVID-19 response, a cooperative agreement between the National Institute for Communicable Diseases of the National Health Laboratory Service and the United States Centers for Disease Control and Prevention (grant number 5 U01IP001048-05-00); the African Society of Laboratory Medicine (ASLM) and Africa Centers for Disease Control and Prevention through a sub-award from the Bill and Melinda Gates Foundation grant number INV-018978; the UK Foreign, Commonwealth and Development Office and Wellcome (Grant no 221003/Z/20/Z); the South African Medical Research Council (Reference number SHIPNCD 76756); the Department of Health and Social Care and managed by the Fleming Fund and performed under the auspices of the SEQAFRICA project; German Federal Ministry of Education and Research (BMBF; grant number 01KA1606; and G7 collaboration grant with the Robert Koch Institute for COVID19) for the African Network for Improved Diagnostics, Epidemiology and Management of common infectious Agents (ANDEMIA). P.L.M. is supported by the South African Research Chairs Initiative of the Department of Science and Innovation and the NRF (Grant No 98341) and the Strategic Health Innovations Program of the SA MRC.
Footnotes
↵* joint first authors;
A typo was corrected in the abstract. N484K, corrected to E484K. Also spelling of corresponding author corrected from Jinal Bhiman to Jinal N. Bhiman.
References



