
Abstract
UK Biobank has released the whole-exome sequencing (WES) data for 200,000 participants, but the best practices remain unclear for rare variant tests, and an existing approach, SAIGE-GENE, can have inflated type I error rates with high computation cost. Here, we propose SAIGE-GENE+ with greatly improved type I error control and computational efficiency compared to SAIGE-GENE. In the analysis of UKBB WES data of 30 quantitative and 141 binary traits, SAIGE-GENE+ identified 551 gene-phenotype associations. In addition, we showed that incorporating multiple MAF cutoffs and functional annotations can help identify novel gene-phenotype associations and SAIGE-GENE+ can facilitate this.
Main
UK Biobank (UKBB) recently released the whole-exome sequencing (WES) data for 200,000 participants1, allowing for studying rare variant associations for complex traits and diseases. However, the best practices remain unclear for set-based rare variant tests in large-scale biobanks. A common practice is to test for associations with all rare (minor allele frequency, MAF ≤ 1%,) loss-of-function (LoF) and missense variants, but this approach can lose power if association signals are enriched in very rare variants or certain functional annotation classes. To improve power, researchers can restrict the test for more rare variants such as variants with MAF ≤ 0.1% or MAF ≤ 0.01%. Another approach is incorporating functional annotations. Since multiple variant sets with different MAF cutoffs and functional annotations exist, tests should be done multiple times for each gene or region and results should be aggregated using minimum p-value or Cauchy combinations2,3.
Currently, SAIGE-GENE4 is the only method developed to conduct the set-based rare variant tests for binary phenotypes with unbalanced case-control ratios in biobank-scale data. For example, in our evaluation, the most recent set-based test, STAAR2, cannot control for type I error rates in the presence of case-control imbalance (Supplementary Figure 1). SAIGE-GENE has been recently used for exome-wide association analysis for 3,700 phenotypes of 281,850 individuals in the UKBB WES data5. In the evaluation of the method using the UKBB WES data with 160K white British samples, we have found that all the tests (Burden, SKAT6 and SKAT-O7) in SAIGE-GENE performed well when testing all rare variants with MAF ≤ 1% (Figure 1A), but inflation was observed in SKAT and SKAT-O tests in SAIGE-GENE when restricting to variants with MAF ≤ 0.1% or MAF ≤ 0.01% and the case-control rates were more unbalanced than 1:30 (Figure 1A, Supplementary Figure 2). To examine whether the inflation is due to inflated type I error rates or polygenicity of the phenotypes, we carried out type I error simulation studies with various case-control ratios (Methods), and observed the same inflation (Supplementary Figure 3). This suggests that SKAT and SKAT-O in SAIGE-GENE can suffer from inflated type I error rates.

The tests were performed for 18,372 genes with missense and loss-of-function (LoF) variants with three different maximum MAF cutoffs: 1%, 0.1%, and 0.01%. Names of genes reaching the exome-wide significant threshold (p-value < 2.5×10−6) in SAIGE-GENE+ are annotated in the plots.
A. SAIGE-GENE B. SAIGE-GENE+
In addition, the computation cost is not low enough to test for the multiple variant sets. For example, to test the largest gene (TTN) with 16,227 variants in the UKBB WES data with 3 different MAF cutoffs (MAF ≤ 1%, 0.1%, 0.01%) and 3 different annotations (LoF only, LoF+missense, and LoF+missense+synonymous), SAIGE-GENE required 197 CPU hours and 65 Gigabytes (Gb) of memory (Supplementary Table 1).
To address these issues, we propose SAIGE-GENE+. Although SAIGE-GENE uses various approaches, including saddlepoint approximation and exact resampling, to adjust for unbalanced case-control ratios, these approaches cannot fully address the imbalance and sparsity in the data (Figure 1A, Supplementary Figure 3A). In order to reduce the sparsity, prior to the set-based association tests, SAIGE-GENE+ collapses ultra-rare variants with MAC ≤10 in the ”absence and presence” way that has been commonly adapted in the association analysis of ultra-rare variants8 by assuming those variants have the same effect direction on the testing phenotype (Methods). We observed that the inflation of SKAT and SKAT-O has been substantially reduced in SAIGE-GENE+ and all tests have well controlled type I errors in both simulated (Supplementary Figure 3B) and the UKBB WES data (Figure 1B) for four exemplary phenotypes with case-control ratios 1:32 to 1:267. The genomic control inflation factors also became closer to one (Supplementary Figure 2).
Collapsing ultra-rare variants in SAIGE-GENE+ decreases the number of variants in each gene or region (Supplementary Figure 4), leading to reduced computation time and memory usage (Figure 2A, Supplementary Table 1). To further reduce computational cost, SAIGE-GENE+ uses a sparse matrix to store dosages or genotypes of the tested variant sets. The computation time of SAIGE-GENE+ for conducting all Burden, SKAT, and SKAT-O was 210 times decreased (9,851 mins vs. 47 mins) and the memory usage dropped from 65Gb to 2.8Gb compared to SAIGE-GENE when testing the largest gene TTN (16,227 LoF+missense+synonymous variants) for its association with the basal metabolic rate (Supplementary Table 1). The overall computation time for testing 18,372 genes with 150,000 samples using three MAF cutoff 1%, 0.1%, and 0.01% and three different variant annotations: LoF only, LoF+missense, and LoF+missense+synonymous, required 1118 CPU hours and 3.9Gb memory (Supplementary Figure 5).

{kind=link}
{kind=link}
A. Computation time and memory of the gene-based tests (Step2, See Method) in SAIGE-GENE and SAIGE-GENE+ for four example genes with different number of variants. The SKAT-O tests were conducted with three maximum MAF cutoffs: 1%, 0.1%, and 0.01% and three variant annotations: LoF only, LoF + missense, and LoF + missense +synonymous and combined using the Cauchy combination. The plots are in the log10-log10 scale. The details of the numbers and genes are presented in the Supplementary Table 1.
B. Most significant variant sets across the three different MAF cutoffs: ≤1%, ≤ 0.1%, and ≤ 0.01% and three functional annotations: LoF (L) only, LoF + missense (M+L), and LoF + missense +synonymous (S+M+L). Distribution of variant sets with the smallest p-values, among 551 significant gene-phenotype associations identified by SAIGE-GENE+ in the analyses of 30 quantitative traits and 141 binary traits in the UKBB WES data.
By collapsing ultra-rare variants, SAIGE-GENE+ can have more significant p-values for several well-known gene-phenotype associations than SAIGE-GENE, even though the later has inflated type I errors. We applied SAIGE-GENE and SAIGE-GENE+ to 37 self-reported binary phenotypes in the UKBB WES data using three different maximum MAF cutoffs: 1%, 0.1%, and 0.01% to 18,372 gene including all missense and LoF variants. We have observed 27 SKAT-O tests with more significant p-values in SAIGE-GENE+ than in SAIGE-GENE (Supplementary Table 6). For example, BRCA2 for breast cancer with MAF ≤ 0.1% has p-value 7.62×10−8 in SAIGE-GENE+ and 1.65×10−3 in SAIGE-GENE, and GCK for diabetes with MAF ≤ 0.1% p-value 1.22×10−13 in SAIGE-GENE+ and 4.06×10−6 in SAIGE-GENE.
We applied SAIGE-GENE+ to analyze 18,372 genes in the UKBB WES data using three different MAF cutoffs: 1%, 0.1%, and 0.01% and three different variant annotations: LoF only, LoF+missense, and LoF+missense +synonymous for 30 quantitative and 141 binary traits. The nine SKAT-O test p-values were then combined using Cauchy combination or the minimum p-value approach (Methods). 465 gene-phenotype associations were significant at the exome-wide significance threshold (p-value ≤2.5×10−6) for 27 quantitative traits (Supplementary Table 2) and 86 gene-phenotype associations for 51 binary traits (Supplementary Table 3). Since the expected number of p-values < 2.5×10−6 under no association across all the phenotypes is 7.85, the false discovery rate is 0.014. Known genes BRCA1, BRCA2, CHEK2, PALB2, and SAMHD1 were significant for breast cancer, CNTNAP3B, CDKN2A and MITF for melanoma, IL33 for asthma, GCK for type 2 diabetes, and LDLR for high cholesterol and ischemic heart disease. For quantitative traits, MC4R and GIPR were significant for body mass index, CETP, LIPG, and LPL for the HDL cholesterol, LDLR and PCSK9 for LDL, and MEPE for heel bone mineral density. We also identified potentially novel gene-phenotype associations. For binary traits, a pancreatic cancer susceptibility gene NOC2L9 was significant for hypovolemia (p-value = 9.68×10−7), CYP21A2, known for the 21-hydroxylase deficient congenital adrenal hyperplasia (CAH)10, for allergy/adverse effect of penicillin (p-value= 3.63×10− 7). Also for quantitative traits, CHEK2, known to be associated with breast cancer and sex hormone-binding globulin measurement11, was significant for the age at menopause (p-value = 4.51×10−17), IGLL5, which encodes an immunoglobulin lambda-like polypeptide, for the lymphocyte count (p-value = 8.06×10−10), and NANOG, which mediates germline development12 and is highly expressed in embryonic carcinoma13, for age at first live birth (p-value = 2.01 ×10−6).
Including lower MAF cutoff variant sets helped identify novel associations. For example, the association between PDCD1LG2, which encodes Programmed Cell Death 1 Ligand, and chronic lymphoid leukemia became significant when tests were restricted to variants with MAF ≤ 0.01% and 0.1% (p-value = 7.5×10−7) compared to testing all variants with MAF ≤ 1% (p-value= 5.4×10−4) (Supplementary Table 4). In addition, including lower MAF cutoff sets helped to replicate known associations including MLH1 for colorectal cancer and CDKN2A for melanoma (Supplementary Table 4). Due to the multiple comparison burden, including lower MAF cutoff sets can make marginally significant associations insignificant. For 141 binary phenotypes, 17 out of 92 (18.4%) associations were further identified with lower MAF cutoff sets, while 9 (9.8%) associations became insignificant (Supplementary Figure 6A, Supplementary Table 4). For 30 quantitative traits, 28 out of 465 (6%) associations were further identified, while 53 (11.4%) associations became insignificant (Supplementary Figure 6A, Supplementary Table 5), suggesting that restricting association tests to rarer variants has a gain for binary phenotypes. In functional annotation categories, 184 associations were identified when the tests were conducted to LoF variants only, including LoF+missense sets identified 299 additional associations, and when LoF+missense+synonymous sets were also included, 91 more associations were identified (Supplementary Figure 6B). We also investigated among the 551 significant gene-phenotype associations, which variant set had the smallest p-value (Figure 2B). Interestingly, among sets with MAF ≤0.01%, LoF variant sets generally had the smallest p-values, while when the MAF cutoff ≤ 1%, LoF+missense+synonymous sets generally had the smallest p-values.
In summary, our results demonstrated that with incorporating multiple MAF cutoffs and functional annotations the exome-wide rare-variant association tests can help identify novel gene-phenotype associations and our SAIGE-GENE+ can facilitate this. In addition, SAIGE-GENE+ has an option to allow users to fit the null generalized linear mixed model with a sparse genetic relationship matrix (GRM) to further reduce the computation burden (Supplementary Note and Supplementary Figure 7 to 10).
Methods
Collapsing ultra-rare variants
Ultra-rare variants with MAC ≤10 were collapsed in the ”absence and presence” way8. More specifically, all ultra-rare variants were collapsed to a new variant, whose genotype was assigned as the maximum genotype value among all ultra-rare variants.
Aggregating multiple tests
For each gene or region, p-values of multiple testing sets corresponding to multiple maximum MAF cutoffs and functional annotations were aggregated using Cauchy combinations2,3. Note that Cauchy combination does not work when any p-value is 1. Thus, we used the minimum p-value approach to combine multiple tests when any test has p-value = 1.
Type I error evaluation
To evaluate the type I error control of SAIGE-GENE and SAIGE-GENE+, we simulated binary phenotypes under the null hypothesis of no genetic effects based on the observed genotypes by WES of the 166,955 samples with white British ancestry in the UK Biobank. The phenotypes were simulated based on real genotypes of randomly selected L= 30,000 LD-pruned (r2 < 0.2) markers from the odd chromosomes with MAF ≥1% from the following logistic mixed model 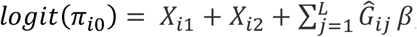 , where πi0 is the probability for the ith individual being a case given covariates and random effects,
, where πi0 is the probability for the ith individual being a case given covariates and random effects, 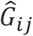 the standardized genotype value for the jth marker of ith individual, and β is the genetic effect size following N(0, τ/L), where τ = 1, which is the variance component parameter. Two covariates, Xi1 and Xi2, were simulated from Bernoulli(0.5) and N(0,1). The intercept α0was determined by given prevalence (i.e. case-control ratios). We repeated the simulation for 20 times for different disease prevalence: 0.3%, 1%, and 10%, respectively. For each phenotype set, a null logistic mixed model was fitted in Step 1 with covariates including the first 4 genetic principal components, which were estimated for all White-British participants in the UK Biobank, X1and X2. Then following each model fitting, in Step 2, we conducted gene-based tests for 7,932 genes on the even chromosomes with missense and LoF variants using three different maximum MAF cutoffs: 1%, 0.1%, and 0.01%. In total, 158,640 gene-based tests were conducted for each maximum MAF cutoff for SAIGE-GENE and SAIGE-GENE+, respectively, and the Q-Q plots were shown in Supplementary Figure 3. Our simulation results suggest that SAIGE-GENE+ has well controlled type I errors with case-control ratios < 1:100 when testing variants with maximum minor allele frequency (MAF) = 0.01% (Supplementary Figure 3B).
the standardized genotype value for the jth marker of ith individual, and β is the genetic effect size following N(0, τ/L), where τ = 1, which is the variance component parameter. Two covariates, Xi1 and Xi2, were simulated from Bernoulli(0.5) and N(0,1). The intercept α0was determined by given prevalence (i.e. case-control ratios). We repeated the simulation for 20 times for different disease prevalence: 0.3%, 1%, and 10%, respectively. For each phenotype set, a null logistic mixed model was fitted in Step 1 with covariates including the first 4 genetic principal components, which were estimated for all White-British participants in the UK Biobank, X1and X2. Then following each model fitting, in Step 2, we conducted gene-based tests for 7,932 genes on the even chromosomes with missense and LoF variants using three different maximum MAF cutoffs: 1%, 0.1%, and 0.01%. In total, 158,640 gene-based tests were conducted for each maximum MAF cutoff for SAIGE-GENE and SAIGE-GENE+, respectively, and the Q-Q plots were shown in Supplementary Figure 3. Our simulation results suggest that SAIGE-GENE+ has well controlled type I errors with case-control ratios < 1:100 when testing variants with maximum minor allele frequency (MAF) = 0.01% (Supplementary Figure 3B).
In addition, to evaluate the type I error control of SAIGE-GENE, SAIGE-GENE+ and STAAR (Figure 1, Supplementary Figure 1) in real data, we applied the methods to four exemplary self-reported binary phenotypes with various case control ratios in the UKBB WES data to 18,372 genes including all LoF and missense variants using three different maximum MAF cutoffs: 1%, 0.1%, and 0.01%. For STAAR, we used the relative coefficient cutoff 0.05 for the sparse GRM to fit the null models.
UK Biobank WES data analysis
We applied SAIGE-GENE+ to analyze 18,372 genes in the UKBB WES data of 166,955 white British samples for 30 quantitative traits and 141 binary traits. Three different maximum MAF cutoffs: 1%, 0.1%, and 0.01% and three different variant annotations: LoF only, LoF + missense, and LoF + missense +synonymous were applied followed by aggregating the multiple SKAT-O tests using Cauchy combination2,3 for each gene. Variants were annotated using ANNOVAR14. The LoF variants include those annotated as frameshift deletion, frameshift insertion, non-frameshift deletion, non-frameshift insertion, splicing, stop gain, and stop loss. Sex, age when attended assessment center, and first four PCs that were estimated using all samples with White British ancestry were adjusted in all tests. 250,656 pruned markers with MAF ≥1%, which were pruned from the directly genotyped markers using the following parameters, were used to construct GRM: window size of 500 base pairs (bp), step-size of 50 bp, and pairwise r2 < 0.2. We used the relative coefficient cutoff 0.05 for the sparse GRM for the variance ratio estimation after fitting the null models. The model was fitted with leave-one-chromosome-out (LOCO) to avoid the proximal contamination.
Computation evaluation
SAIGE-GENE and SAIGE-GENE+ use a two-step approach. Step1 estimates the model parameters (i.e. variance component and fixed effect coefficients) in the null model and Step2 conducts gene-based association tests. Since both SAIGE-GENE and SAIGE-GENE+ use the same approach in Step1, we only compared computation time and memory usages of Step2 (Figure 2A, Supplementary Table 1, Supplementary Figure 5). Note that model parameters need to be estimated only once for each phenotype and can be used genome-wide regardless of MAF cutoffs and functional annotations. The computation cost of Step1 in SAIGE-GENE+ was given in Supplementary Figure 10. SAIGE-GENE+ has an option to use a sparse genetic relationship matrix (GRM), which further reduces computation cost in Step1 (Supplementary Note).
Code and data availability
SAIGE-GENE+ is implemented as an open-source R package available at https://github.com/weizhouUMICH/SAIGE/master. The summary statistics and QQ plots for 30 quantitative phenotypes and 141 binary phenotypes in UK Biobank by SAIGE-GENE+ are currently available for public download at https://storage.googleapis.com/leelabsg/saige-gene/reformat_all_withPhenoDetails.txt
Competing financial interests statement
B.M.N. is a member of Deep Genomics Scientific Advisory Board, has received travel expenses from Illumina, and also serves as a consultant for Avanir and Trigeminal solutions. K.J.K is a consultant for Vor Biopharma.
Author contributions
W.Z., W.B., Z.Z., and S.L. designed experiments. W.Z., W.B., Z.Z., performed experiments and analyzed the UK Biobank data. W.Z. implemented the software with input from W.B. and Z.Z.. Helpful advice was provided by K.K.D, K.A.J., K.J.K., B.M.N., and M.J.D.. W.Z., W.B., Z.Z., and S.L. wrote the manuscript with input from all co-authors.
Acknowledgments
This research has been conducted using the UK Biobank Resource under application number 45227. SL was supported by Brain Pool Plus (BP+, Brain Pool+) Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (2020H1D3A2A03100666, S.L). WB and ZZ were supported by NIH R01 HG008773. WZ was supported by the National Human Genome Research Institute of the National Institutes of Health under award number T32HG010464. We thank Dr. Alkes Price for the constructive comments and suggestions.
References




Subject Area
Reviews and Context
0
Comment
0
TRIP Peer Reviews
0
Community Reviews
0
Automated Services
0
Blogs/Media
Author Videos