
ABSTRACT
The transmission networks of SARS-CoV-2 in sub-Saharan Africa remain poorly understood. We analyzed 684 genomes from samples collected across six counties in coastal Kenya during the first two waves (March 2020 -February 2021). Up to 32 Pango lineages were detected in the local sample with six accounting for 88.0% of the sequenced infections: B.1 (60.4%), B.1.1 (8.9%), B.1.549 (7.9%), B.1.530 (6.4%), N.8 (4.4%) and A (3.1%). In a contemporaneous global sample, 571 lineages were identified, 247 for Africa and 88 for East Africa. We detected 262 location transition events comprising: 64 viral imports into Coastal Kenya; 26 viral exports from coastal Kenya; and 172 inter-county import/export events. Most international viral imports (61%) and exports (88%) occurred through Mombasa, a key coastal touristic and commercial center; and many occurred prior to June 2020, when stringent local COVID-19 restriction measures were enforced. After this period, local transmission dominated, and distinct local phylogenies were seen. Our analysis supports moving control strategies from a focus on international travel to local transmission.
INTRODUCTION
By 15th June 2021, at least 175 million cases of coronavirus disease 2019 (COVID-19) and > 3.7 million associated deaths were reported worldwide (1). By the same date, Kenya reported 175,176 laboratory-confirmed cases of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and 3,396 COVID-19 associated deaths (2). Serological surveys indicated that Kenya’s COVID-19 epidemic had progressed further than could be discerned from the limited laboratory testing case reports that were available during the period (3, 4). At the end of Kenya’s first wave (August 2020), nationwide anti-SARS-CoV-2 IgG prevalence based on analysis of blood donor samples was estimated to be 9% (5).
By June 2021, Kenya had experienced three waves of SARS-CoV-2 infections. The first wave peaked in July-August of 2020, the second in November 2020 and the third in March-April 2021 (6). Despite this progression, the local SARS-CoV-2 spread patterns remain poorly understood. The analysis presented here examined genome sequences from the first two waves in Coastal Kenya. Overall, the documented SARS-CoV-2 infections in Kenya during these early two waves were concentrated in cities, especially Nairobi (∼42%), the capital, and Mombasa (∼8%), the country’s second largest city on the Indian ocean coast. In addition to Mombasa, there are five other counties that make up Kenya’s Coastal region namely, Kilifi, Kwale, Taita Taveta, Tana River and Lamu. The region is a major tourism destination accessible through multiple airports, seaports and land border entry points from Tanzania through Kwale and Taita Taveta counties (7).
Throughout the COVID-19 pandemic, genomic surveillance has been critical for tracking the spread of SARS-CoV-2 and investigating viral evolution (8-11). In Kenya, genome sequencing started soon after the identification of the initial case on 13th March 2020 and has remained a key tool informing public health response decisions. We have previously reported on the genomic analysis of SARS-CoV-2 in Coastal Kenya during the early phase of the pandemic (up to July 2020) revealing multiple lineage introductions but with mainly lineage B.1 establishing local transmission (7). Here we report the genetic composition of the subsequent two waves (up to February 2021) with the aim of documenting patterns of viral introductions, evolution and spread across the six Coastal counties of Kenya.
RESULTS
Infection waves in Coastal Kenya counties
The first cases of COVID-19 in Coastal Kenya were reported in Mombasa and Kilifi counties in March 2020, Figure 1A. The timing of the first infection peaks across the six counties differed, with Mombasa showing the earliest surge. By 26th February 2021, Mombasa, Lamu and Taita Taveta had observed two major infection peaks while Kilifi, Kwale and Tana River had experienced only a single peak, with the latter two documenting very few cases, Figure 1A. During this period, the Kenya Ministry of Health (MoH) reported a total of 12,307 laboratory confirmed SARS-CoV-2 cases for the six Coastal counties: Mombasa (n=8,450, 68.7%), Kilifi (n=2,458, 20.0%), Taita Taveta (n=567, 4.6%), Kwale (n=437, 3.6%), Lamu (n=309, 2.5%) and Tana River (n=85, 0.7%), S1 Figure.
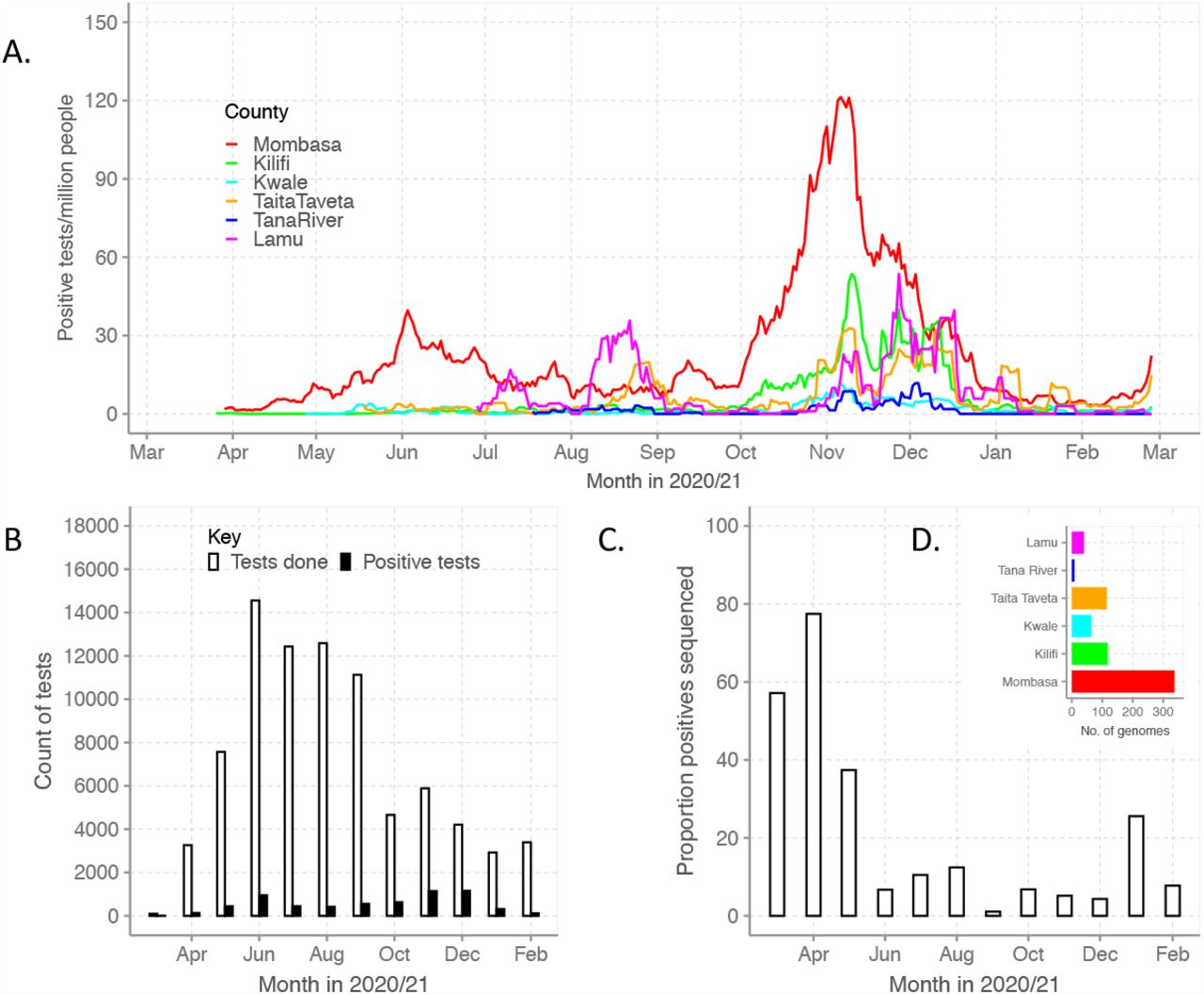
Panel A, epidemic curves for each of the six Coastal Kenya counties derived from the daily positive case numbers reported by the Ministry of Health. Panel B, monthly count of SARS-CoV-2 RT-PCR tests undertaken at the KEMRI-Wellcome Trust Research Programme (KWTRP) and those positive during the study period. Panel C, monthly proportion of samples sequenced from total SARS-CoV-2 positives detected at KWTRP. Panel D, Distribution of the sequenced 684 samples across the 6 Coastal Kenya counties.
SARS-CoV-2 testing and sequencing at KWTRP
The KEMRI-Wellcome Trust Research Programme (KWTRP) was designated as the government SARS-CoV-2 testing center for Coastal region soon after identification of the first case in Kenya. Between 17th March 2020 and 26th February 2021, we tested 82,716 nasopharyngeal/oropharyngeal (NP/OP) swab samples from the six Coastal counties and 6,307 (7.6%) of these were determined as SARS-CoV-2 positive (approximately half of the total reported by MoH), distributed by month as in Figure 1B. Overall, a total of 3,137 (6.8%) tests at KWTRP were positive from samples taken in Mombasa, 1,142 (8.8%) for Kilifi, 657 (4.5%) for Taita Taveta, 349 (12.7%) for Lamu, 437 (7.9%) for Kwale and 105 (12.0%) for Tana River.
We sequenced the genomes of 684 (10.8%) RT-PCR positive samples as follows: Mombasa: 337 (10.7%), Kilifi: 118 (8.1%), Taita Taveta: 115 (13.4%), Kwale: 65 (14.9%), Lamu: 40 (11.5%) and Tana River: 9 (8.5%), Figure 1C and 1D. The sequences were spread across the study period with 272 (39.8%) from the introduction phase (17th March to 20th May 2020 when nationally <50 new cases were reported per day), 169 (24.7%) from wave one (21st May-15th September 2020) and 243 (35.5%) from wave two (16th September 2020 to 26th February 2021), S1 Figure. Overall, our sequencing rate corresponded to approximately one sequence for every 18 confirmed cases.
Lineage dynamics in Coastal Kenya
The 684 genomes were classified into 32 Pango lineages, including four first identified in Kenya (N.8, B.1.530, B.1.549 and B.1.596.1), S1 Table. During the introductions phase we detected 12 lineages while during wave one we detected 16 lineages (11 of which were being identified locally for the first time) and during wave two we detected 18 lineages (nine of which were being identified locally for the first time). Thirteen lineages (40.6%) were identified in three or more samples with the top six accounting for 88.0% of the sequenced infections: B.1 (n=413, 60.4%, European lineage which predominated in the Northern Italy outbreak early in 2020), B.1.1 (n=61, 8.9%, European lineage), B.1.549 (n=54, 7.9%, Kenyan lineage), B.1.530 (n=44, 6.4%, Kenyan lineage), N.8 (n=30, 4.4%, Kenyan lineage) and A (n=21, 3.1%, lineage root to the pandemic first observed in China).
Lineage B.1 was the first to be detected in the region and comprised the initial cases identified in Mombasa, Kilifi, Kwale and Tana River, Figure 2. This lineage comprised 71.3% (n=194) of the viruses we sequenced from the introduction phase, 65.7% (n=111) from wave one and 44.4% (n=108) from wave two. Overall, B.1 accounted for most of the virus sequences detected in all counties except Lamu where lineage N.8 (alias lineage B.1.1.33.8) was most frequent (70% of the cases, first detected there on 23rd June 2020). Only two additional sequenced cases of Lineage N.8 occurred outside Lamu County (one in Mombasa and another in Kilifi).

Lineages detected in less than three cases were put together and referred to as “other lineages”. This group included 18 lineages namely: A.25, B.1.1.254, B.1.179, B.1.378, B.1.393, B.1.1.111, B.1.1.33, B.1.1.485, B.1.1.7, B.1.1.70, B.1.153, B.1.340, B.1.450, B.1.526, B.1.535, B.1.553, B.1.558 and B.4 (see details in S1 Table).
Although lineage A was also detected in Mombasa and Taita Taveta, most of its cases occurred in Kwale where it comprised 20% of all cases identified in the county, Figure 2. Lineage B.1.1 was mostly observed in Mombasa with limited number of cases observed in Kilifi, Kwale and Taita Taveta.
During the wave two period in Coastal Kenya, there was an emergence of three lineages first identified in Kenya: (a) lineage B.1.549 (first observed 26th June 2020) which was responsible for a significant number of the sequenced infections in Kilifi (n=24), Mombasa (n=18), Taita-Taveta (n=5), and Kwale (n=7), (b) lineage B.1.596.1 (first observed 01st August 2020) and detected in Mombasa, Kilifi, Kwale and Taita-Taveta and (c) lineage B.1.530 (first observed 28th October 2020) that predominated infections in Taita-Taveta (n=28) with a fewer cases observed in Mombasa (n=8), Kilifi (n=7) and Lamu (n=1).
Two known variants of concern (VOC) were detected during the study period: the lineage B.1.351 (the Beta variant in 10 samples; and the lineage B.1.1.7 (the Alpha variant in one sample (12). The earliest B.1.351 infections were identified in international travelers from South Africa in mid-December 2020 while the earliest detected B.1.1.7 case was in second week of January 2020 in a local with no history of recent international travel. We also detected three cases each of A.23 and A.23.1 after the peak of wave two (both lineages first identified in Uganda, and are considered variants of interest (VOI) in the region (13)).
Lineage dynamics with a widening scale of observation
We compared the temporal prevalence patterns of the lineages identified in Coastal Kenya to Eastern Africa, Africa and a global sub-sample, Figure 3. In these comparative genome sets, we identified 88 Pango lineages for Eastern Africa, 247 for Africa and 571 for the global sub-sample.

Panel A, Biweekly distribution of detected lineages in Coastal Kenya from the sequenced 684 genomes. Panel B, biweekly distribution of detected lineages in Eastern Africa from 2,316 contemporaneous genomes from 10 countries whose contemporaneous data were available in GISAID. Lineages not observed in the Coastal Kenya dataset have been lumped together as “Absent Kenya” but with the those appearing among top six and not observed in Kenya indicated just below the other Kenya group. Panel C, weekly distribution of detected lineages in Africa from 12,506 contemporaneous genomes from 39 countries that were available in GISAID. Panel D, weekly distribution of detected lineages in a global sub-sample of 9,989 contemporaneous genomes from 112 countries that were compiled from GISAID (see detail in methods section).
In the Eastern Africa sample (n=2,315), all the top lineages were also observed in the Coastal Kenya data (B.1.351, A.23.1, B.1, B.1.1, B.1.177) except lineage B.1.380 (n=128, a Rwanda lineage), Figure 3B. In the Africa sample (n=12,506), four of the six top lineages (B.1, B.1.1, B.1.351 and B.1.1.7) were also identified in Coastal Kenya. The undetected two lineages were B.1.1.448 (n=392) and C.1 (n=330) both of which were mainly observed South Africa samples. In the global sub-sample (n=8,909) four of the top six lineages (B.1, B.1.1, B.1.1.7 and B.1.177) where observed in the samples from Coastal Kenya. The unobserved lineages were D.2 (Alias of B.1.1.25.2, an Australian lineage) and B.1.1.28 (a Brazilian lineage).
At the time of the second wave in Kenya, the VOC (B.1.1.7 and B.1.351) were already widespread across Eastern Africa and Africa but there were only sporadic detections our dataset from Coastal Kenya. Although in the comparison data lineage B.1 occurred in substantial proportions across the different scales early in the pandemic, its prevalence diminished faster overtime in the outside Kenya sample sets when compared to the Coastal Kenya dataset set. Approximately 60% of the lineages comprising infections globally were not seen in the Coastal Kenya samples.
SARS-CoV-2 diversity in Coastal Kenya
When we reconstructed time-resolved ML phylogeny of the Coastal Kenya genomes while including a global reference set of genomes (n=1500) we observed that (i) the Coastal Kenya genomes were represented across many but not all of the major phylogenetic clusters observed globally, ii) some of the coast clusters expanded after introduction whereas others did not (observed as singletons) and iii) all counties appeared to have had multiple variants introduced with some clusters comprising genomes detected across multiple counties, Figure 4A. There was considerable correlation of the root-to-tip genetic distance and the date of sampling (r2=0.442), Figure 4B.
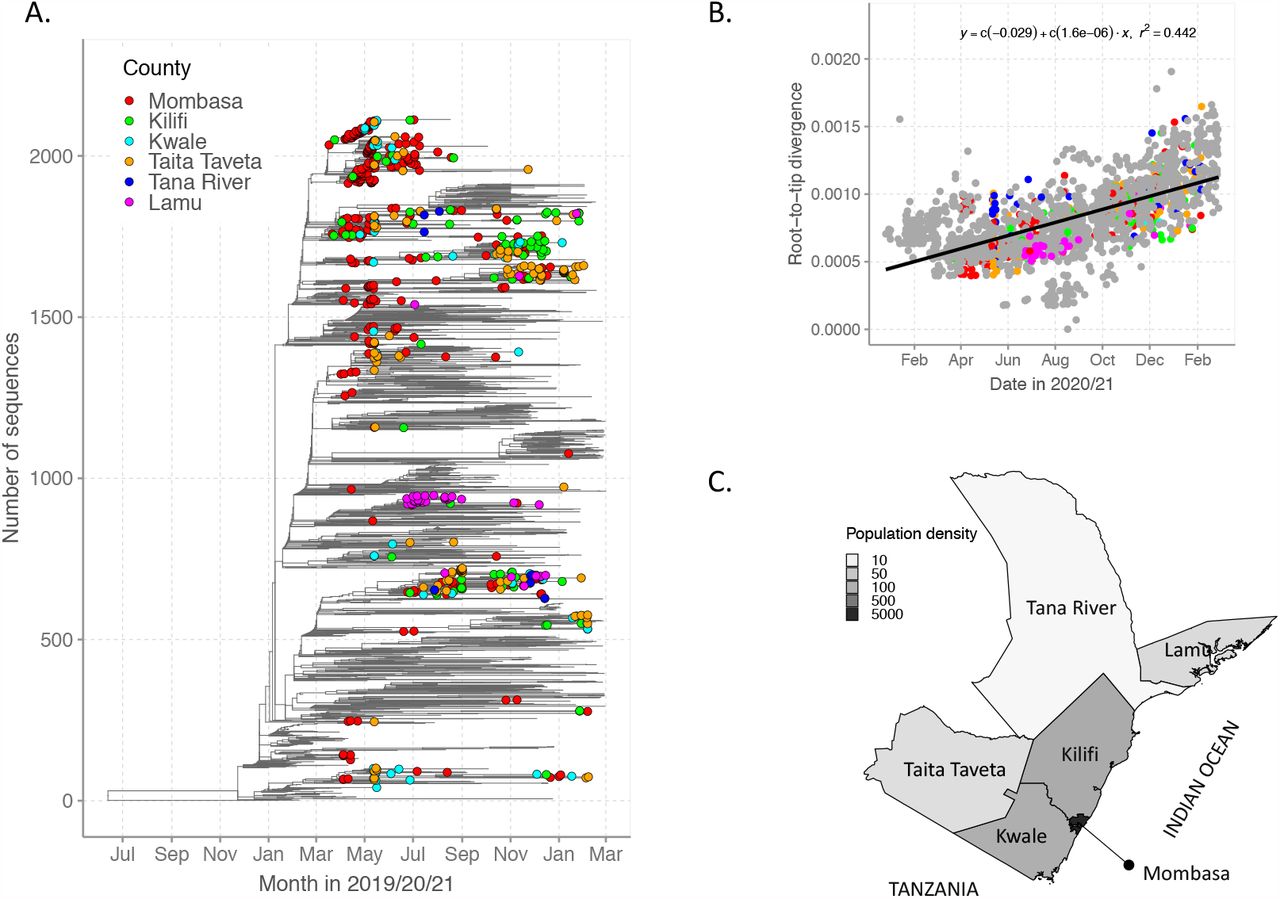
Panel A, Time-resolved global phylogeny that combined 639 Coastal Kenya SARS-CoV-2 genomes and 1474 global reference sequences. Panel B, Roo-to-tip regression analysis coastal Kenya (colored by county) and global (shown in grey) sequences with determination of co-efficient (r2). Panel C, map of the six study counties colored scaled by the population density (number of persons per square kilometer).
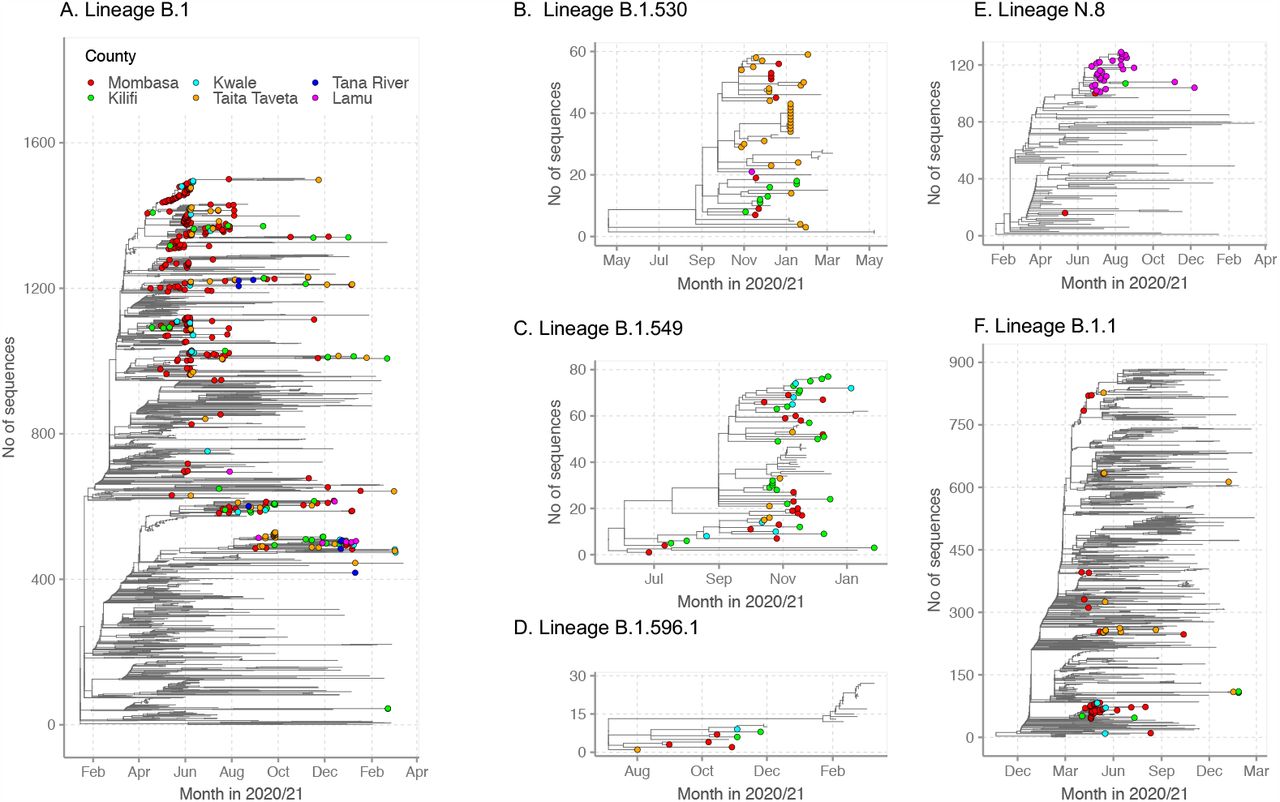
For detailed investigation into the local SARS-CoV-2 genetic diversity, we reconstructed time-resolved lineage specific phylogenetic trees are shown in Figure 5. Viruses occurring within lineage B.1 and lineage B.1.1 showed significant genetic divergence and formed multiple phylogenetic clusters interspersed with other global sequences, a feature indicative of multiple viral introductions into a geographic area. Three lineages first identified in Kenya (B.1.530, B.1.549 and B.1.596.1) were found to (a) possess significant diversity consistent with widescale spread within Kenya (Figure 5B-D), (b) formed multiple county-specific sub-clusters and (c) show local sequences interspersed with global comparison genomes from the same lineage implying potentially export (or import) events, Figure 5C. However, the picture painted by lineage N.8 was different. This lineage was mainly detected in Lamu forming a single monophyletic group (Figure 5E) when co-analysed with its precursor lineage B.1.1.33, an observation consistent with a single introduction then expansion.

Time-resolved lineage-specific phylogenetic trees for the commonly detected lineages in Coastal Kenya. The Coastal Kenya genomes are indicated with filled circles colored by county. Panel A, phylogeny for lineage B.1 that combined 369 Coastal Kenya genomes and 1134 global reference sequences. Panel B, phylogeny for lineage B.1.530 that combined 44 Coastal Kenya genomes and 15 global sequences belonging also to B.1.530. Panel C, phylogeny for lineage B.1.549 that combined 54 Coastal Kenya genomes and 23 sequences from other countries of lineage B.1.549. Panel D, phylogeny for lineage B.1.596.1 combines 8 Coastal Kenya genomes and 19 global reference sequences. Panel E, phylogeny for lineage N.8 that combined 30 Coastal Kenya genomes of lineage N.8, 1 Coastal Kenya genome of lineage B.1.1.33 and 99 global sequences of lineage B.1.1.33. Panel F, phylogeny for lineage B.1.1 that combined 60 Coastal Kenya genomes and 824 global sequences belonging to lineage B.1.1.
Viral imports and export from Coastal Kenya
We used ancestral location state reconstruction of the dated phylogeny (Figure 4) to infer the number of viral import and exports (see details in the methods (14)). In total, between March 2020 and February 2021, we detected 262 location transition events, 64 of which were viral import events into Coastal Kenya from outside and 26 were viral exports events from Coastal Kenya to outside populations, Figure 6A-C. A total of 172 import/export events were detected between the six Coastal counties. Virus imports into the region occurred through Mombasa (n=39, 61%), Kwale (n=10, 16%), Taita Taveta (n=10, 16%), Kilifi (n=3, 5%) and Lamu (n=1, 2%). Virus export from the region occurred through Mombasa (n=23, 88%) and Kwale (12%). Of the 64 detected viral imports from outside the Coastal Kenya region, 40 (63%) occurred during the introductions phase, 12 (19%) occurred during wave one and another 12 (19%) during wave two. Of the 26 detected viral exports, 13 (50%) occurred during introductions phase, 10 (38%) during wave one and three (12%) during wave two, Figure 6B and D.

Panel A, the number of imports and exports of viral lineages from Coastal Kenya shown as an alluvium plot. Panel B, biweekly location transition events observed in the Coastal Kenya and global genomes shown as either viral export, import or inter-county transmission. Panel C, Biweekly number of import events into the individual Coastal counties and their origins.
Amino acid changes in the Coastal Kenya genomes
We scanned the entire SARS-CoV-2 genome for amino acid changes from the Wuhan reference genome in the 684 Coastal Kenya sequences. Below we summarize changes that were identified in ≥5 genomes. We observed 20 changes in the spike (S) protein meeting the set criterion, 15 in the nucleocapsid (N) protein, 14 in ORF3a, 13 in ORF1b, nine in the ORF1a, seven in ORF8, five in ORF6, and one in the envelope (E) protein. No codon changes, meeting of criterion was observed in the matrix (M) protein, ORF7a, ORF7b or ORF10. The most common amino acid changes observed in the Coastal Kenya genomes were S: D614G (94.4%), ORF1b: P314L (92.9%), ORF3a: Q57H (15.5%), ORF1a: T265I (15.4%) and N: R195K (12.9%), S2 Figure.
Several lineage-defining mutations were observed for the Kenya-specific lineages as summarized in S3 Figure.
DISCUSSION
We present evidence of continuous introductions of SARS-CoV-2 into Coastal Kenya documenting at least 64 independent viral introduction events into the region during the first year of the pandemic. Multiple variant introductions were observed even at the individual counties level, with extensive inter-county transmission events after the early phase period, re-affirming that Coastal Kenya populations are quite linked epidemiologically. Strikingly, many of the imports and exports from the region occurred through Mombasa, a major commercial, industrial and tourist destination in the region, before dispersing to the other counties. This observation highlights Mombasa’s central role as a gateway of viral variants into the region and the significance of enhanced and persistent surveillance in this city for early detection of introduced of variants of concern.
The analyzed counties had both differences and similarities in SARS-CoV-2 transmission patterns. While Mombasa, Taita Taveta and Lamu counties had experienced two waves of infections by February 2021, Kilifi, Tana River and Kwale counties had experienced only a single major wave. This may have been brought about by differences in population density, accessibility, and applied infection control measures. For instance, Mombasa is densely populated (∼5,604.64 persons/sq.km, 2019 census), has a large seaport and an international airport while Lamu has a sparsely populated mainland and over 65 islands (total population 143,920, population density, 21.84 persons/sq.km). Taita Taveta and Kwale counties are primarily rural counties bordering Tanzania which had a largely uncontrolled epidemic (15). Tana River is the most remote and sparsely populated (8.9 persons/sq.km) county in the region (Figure 4C).
During the early months of the pandemic several Pango lineages were introduced into the region (A, B.1, B.1.1, B.1.340, B.1.535, B.4 and B.4.7). However, it is lineage B.1 which has a European origin, that became predominant. This lineage has the D614G change in its spike protein considered to enhance fitness (16) and this may have boosted its local transmission and early dominance. Lineage N.8 was specific to Lamu County and B.1.530 to Taita Taveta.
Lineage N.8 has not been observed outside Kenya and its precursor lineage (B.1.1.33) was observed earlier in South America, mainly Brazil. Lineage N.8 may have arisen from a single introduction event into Lamu county, and then remained localized to Lamu. The N.8 lineage has seven characteristic lineage defining mutations including S: D614G and R203K, G204R and I292T on the nucleocapsid protein.
Kilifi county, which neighbors Mombasa to the North (∼1.4 million with a population density of 117.67 persons/sq.km, 2019 census) observed its first peak of infections during the second national wave. The Kilifi first wave was comprised of mainly three lineages: B.1 (53.5%), B.1.530 (8.1%) and B.1.549 (25.6%). Lineages B.1.530 and B.1.549, first identified in Kenya, and may have arisen from local evolution of B.1. Lineage B.1.530 has six characteristic mutations including spike P681H change adjacent to the biologically important furin cleavage site while lineage B.1.549 has seven characteristic mutations, five occurring in the ORF1a or ORF1b. Both two lineages have now been observed in a few other countries albeit in small numbers: seven countries for B.1.530 (Rwanda, Netherlands, Germany, Denmark, USA, Japan and Australia) and three countries for B.1.549 (England, USA, Canada).
Near the tail end of the national wave two, we detected only sporadic cases of the VOC B.1.351 and B.1.1.7. Lineage B.1.351 was first to be observed, initially in Kilifi in two asymptomatic international travelers in mid-December 2020. Lineage B.1.1.7 was subsequently detected in a local who presented to a Mombasa clinic in the second week of January 2021. In the subsequent weeks up to the end of the period covered by this analysis (February 2021), no additional B.1.1.7 was detected unlike lineage B.1.351 which continued to be detected sporadically in January and February 2021, especially in truckers screened at Tanzania border points of entry in Taita Taveta and Kwale counties.
When we compared the local lineage patterns to the global patterns, the VOC were already extensively spread across Eastern Africa (B.1.351 VOC), Africa (B.1.351 VOC) and worldwide (B.1.1.7 VOC) in the last quarter of 2020. This implies that a lag occurred in their arrival into Coastal Kenya perhaps due to public health measures that have remained in place during the period especially at the international borders. It is also notable several lineages were detected globally (>500) but only a small fraction (<10%) of this were documented locally.
Our study contributes to the limited but growing literature illuminating SARS-CoV-2 transmission patterns in Africa (11, 13, 17-19). The patterns revealed have potential to inform pandemic mitigation strategies. The key limitations of our analysis include, first, the samples we sequenced were only those available to us through the rapid response teams (RRTs) whose case identification protocols were altered at the different study phases following guidance from the MoH. Second, we only sequenced ∼10% of the positive samples identified in our laboratory, the majority from the introduction phase and were prioritizing samples with a Ct value of <30.0.
Third, the sampling across the six Coastal counties was not uniform, probably in part due to varied distance from our testing centre located in Kilifi County but also the total number of positive cases varied between the counties.
In conclusion, we show that the first two SARS-CoV-2 waves in Coastal Kenya observed transmission of both newly introduced variants and potentially locally evolved variants. New variant introductions appeared to mainly occur through Mombasa city. Strikingly, only a limited number of the many introduced variants progressed to transmit extensively perhaps due to ongoing public health interventions e.g., screening at ports of entry, case isolation and quarantining of contacts of cases. Unlike in the global contemporaneous sample, we did not find evidence of extensive local transmission of the global VOC during wave two. Thus, we infer that it is more likely that the relaxation of some of the interventions (e.g., reopening of learning institutions, airspace, bars and restaurants) that drove the second wave of infections. Overall, our study shows the importance of detailed local genomic surveillance even in remote and under-resourced settings to understand origins and spread patterns of SARS-CoV-2 to optimize local interventions.
MATERIALS AND METHODS
Ethical statement
Samples analysed here were collected under the MoH protocols as part of the national response to the COVID-19 pandemic. The whole genome sequencing study protocol was reviewed and approved by the Scientific and Ethics Review Committee (SERU), Kenya Medical Research Institute (KEMRI), Nairobi, Kenya (SERU #4035). Individual patient consent was not required by the committee for the use of these samples for genomic surveillance to inform public health response.
Study period and population
This study examined samples collected between 17th March 2020 to 26th February 2021 from six counties in Coastal Kenya i.e Mombasa, Kilifi, Kwale, Tana River, Taita Taveta and Lamu. We divided the study period into three phases based on trends in daily reported COVID-19 national case numbers by the MoH (2): (a) Introduction phase – 17th March-20th May, 2020 (b) Wave one – 21st May-15th September 2020 and (c) Wave two – 16th September to 28th February 2021, S1 Figure. We considered the transition from introduction phase to Wave as the timepoint when the national daily positives exceeded 50 and the transition from Wave one to Wave two as the timepoint when a consistent renewed rise of national daily number of positives started after Wave one peak.
Patient samples
This study analyzed SARS-CoV-2 positive NP/OP swab samples that were collected by the MoH County Department of Health RRTs, across all the six counties of Coastal Kenya. SARS-CoV-2 positive diagnosis was made at the KWTRP in Kilifi County (20). The RRTs delivered the NP/OP swabs to KWTRP laboratories within 48 hours of collection in cool boxes with ice packs. The samples were from persons of any age and sampling followed the MOH eligibility criteria that were revised from time to time (7). Persons sampled included those with acute respiratory symptoms, those with a recent history of travel to the early COVID-19 hotspots, contacts of confirmed cases, persons presenting at international border points seeking entry into Kenya, among other criteria.
SARS-CoV-2 detection at KWTRP
Viral RNA was extracted from the NP/OP samples using any one of seven commercial kits that were available namely, QIAamp Viral RNA Mini Kit, RNeasy ® QIAcube ® HT Kit, QIASYMPHONY ® RNA Kit, T0IANamp Virus RNA Kit, Da An Gene Nucleic acid Isolation and Purification Kit, SPIN X Extraction and RADI COVID-19 detection Kit. The extracts were tested for the presence of SARS-CoV-2 nucleic acid following different protocols depending on which one was available of the following 7 kits/protocols: 1) the Berlin (Charité) primer-probe set (targeting envelope (E) gene, nucleocapsid (N) or RNA-dependent RNA-polymerase (RdRp)), 2) European Virus Archive – GLOBAL (EVA-g) (targeting E or RdRp genes), 3) Da An Gene Co. detection Kit (targeting N or ORF1ab), 4) BGI RT-PCR kit (targeting ORF1ab), 5) Sansure Biotech Novel Coronavirus (2019-nCoV) Nucleic Acid Diagnostic real-time RT-PCR kit or 6) Standard M kit (targeting E and ORF1ab) and 7) TIB MOLBIOL kit (targeting E gene). Protocol-specific recommended cycle threshold cut-offs were followed in defining SARS-CoV-2 positives.
SARS-CoV-2 genome sequencing
Only samples that had a PCR cycle threshold value of <30.0 were targeted for whole genome sequencing (7). Viral RNA from positive samples was reextracted using QIAamp Viral RNA Mini kit following the manufacturer’s instructions and reverse transcribed using LunaScript® RT SuperMix Kit. The cDNA was amplified using Q5® Hot Start High-Fidelity 2x Mastermix along with the ARTIC nCoV-2019 version 3 primers. The PCR products were run on an 1.5% agarose gel and for samples whose SARS-CoV-2 amplification was considered successful were purified using Agencourt AMPure XP beads and taken forward for library preparation. Sequencing libraries were constructed using Oxford Nanopore Technology (ONT) ligation sequencing kit and the ONT Native Barcoding Expansion kit as described in the ARTIC protocol (21). Every MinION (Mk1B) run comprised 23 samples and one negative (no-template) control.
SARS-CoV-2 genome assembly
Following MinION sequencing, the FAST5 files were base-called using the OTN’s software Guppy v3.5-4.2. Consensus SARS-CoV-2 sequences were derived from the reads using the ARTIC bioinformatics pipeline. A threshold of ×20 coverage was required for a base to be included in the consensus genome otherwise it was masked with an N. Only complete or near-complete genomes with >80% coverage and ×20 or more read depth were taken forward for phylogenetic analysis.
Lineage assignment
The consensus genomes were assigned into Pango lineages via the Phylogenetic Assignment of named Global Lineages (PANGOLIN) software suite (Pangolin v2.4.2, Pango v1.1.23) (22). The amino acid changes profiles of the Coastal Kenya genomes from the reference strains was investigated using the nextclade tool v0.14.2 (23).
Global contextual sequences
We prepared three sets of contextual sequences from GISAID deposited as of May 2021. Selected sequences were those with non-ambiguous sampling date, sampled during the period covered by our study and after alignment possessed <1500 ambiguous nucleotides i.e showed >95% genome completeness.
Set 1: We compiled an African SARS-CoV-2 genome set which we assigned Pango lineages and used to compare the continental lineage temporal patterns to the Coastal Kenya lineage distribution. A total of 12,542 genomes were retrieved for this purpose on the 11th of May 2021. From these we also created an Eastern Africa subset which comprised of 2,316 genomes from 10 countries, namely, Zimbabwe, Zambia, Uganda, Rwanda, Renunion (overseas territory), Mozambique, Malawi, Madagascar, Ethiopia and Comoros.
Set 2: We compiled a global reference set of 9,989 genomes collected across all the six inhabited continents between 24th December 2019 and 28th February 2021. Between 23 and 899 genomes were selected for each included month (mean of 667 and median of 701) randomly selected across all continents from across 112 countries. These genomes were used to infer the global temporal patterns of the lineages observed in Coastal Kenya. A subset of these genomes (n=1500) was combined with the Kenyan genomes to infer their global phylogenetic context.
Set 3: We compiled a genotype reference for the top six observed lineages in Coastal Kenya. For lineages B.1. and B.1.1 which >5000 genomes exist in GISAID were prepared we used the sub-sample from Set 2 above. For lineages B.1.530, B.1.549, B.1.596.1 and N.8 we retrieved all genomes assigned these lineages available from GISAID to infer their global phylogenetic context.
Phylogenetic analysis
Multiple sequence alignments were prepared in Nextalign v 0.1.6 software and using the initial Wuhan sequence (Accession number: NC_045512) as the reference. The alignment was manually inspected in SEAVIEW v4.6.4 to spot any obvious misalignments. Quick non-bootstrapped neighbor joining trees were created in SEAVIEW to identify any aberrant sequences that were henceforth discarded.
We reconstructed maximum likelihood (ML) phylogenies using IQTREE v2.1.3. The software initiates tree reconstruction after assessment and selection of the best model of nucleotide substitution for the alignment. The ML trees were linked to the various metadata in R programming software v4.0.2 and visualized using R ggTree v2.4.2. TempEst v1.5.3 was used to assess the presence of a molecular clock signal in analysed data and linear regression of root-to-tip genetic distances against sampling dates plotted.
Import/export analysis
The global ML tree topology was used to estimate the number of viral transmission events between Coastal Kenya and the rest of the world as in (17). The software TreeTime was used to transform the ML tree topology into a dated tree assuming a constant genomic evolutionary rate of SARS-CoV-2 of 8.0 ×10−4 nucleotide substitutions per site per year (14). Outlier sequences were identified by TreeTime and excluded during this process. A migration model was henceforth fitted to the resulting time-scaled phylogenetic tree from TreeTime, mapping the location status of the genomes from the six counties at both the tips and internal nodes. Using the date and location annotated tree topology, we counted the number of transitions between and within Coastal Kenya counties and the rest of the world and plotted this using ggplot2 v3.3.3.
Epidemiological data
The Kenya daily case data for the period between March 2020 and February 2021 was downloaded from Our World in Data database (https://ourworldindata.org/coronavirus/country/kenya). Metadata for the Coastal Kenya samples was compiled from the MoH case investigation forms.
Kenya COVID-19 response
We derived status of Kenya government COVID-19 interventions using data from Our World in Data database that has calculated the Oxford Stringency Index (SI). These SI estimations are composite measure based on nine response indicators implemented by governments including school closures; workplace closures; cancellation of public events; restrictions on public gatherings; closures of public transport; stay-at-home requirements; public information campaigns; restrictions on internal movements; and international travel controls, rescaled to a value 0-100, with 100 being strictest (24). The Kenya government revised the policy interventions at approximately monthly intervals (6) and the changes in SI overtime are shown in S1 Figure.
Data Availability
Coastal Kenya genomes have been deposited to both GISAID (Accession numbers: EPI_ISL_1039223-29; EPI_ISL1440075-109, IPI_ISL_457845-931; EPI_ISL_568695-872; EPI_ISL806548-717; EPI_ISL_855494-548; EPI_ISL_968807-9245).
Members of COVID-19 Testing Team at KWTRP
Agnes Mutiso, Alfred Mwanzu, Angela Karani, Bonface M. Gichuki, Boniface Karia, Brian Bartilol, Brian Tawa, Calleb Odundo, Caroline Ngetsa, Clement Lewa, Daisy Mugo, David Amadi, David Ireri, Debra Riako, Domtila Kimani, Edwin Machanja, Elijah Gicheru, Elisha Omer, Faith Gambo, Horace Gumba, Isaac Musungu, James Chemweno, Janet Thoya, Jedida Mwacharo, John Gitonga, Johnstone Makale, Justine Getonto, Kelly Ominde, Kelvias Keter, Lydia Nyamako, Margaret Nunah, Martin Mutunga, Metrine Tendwa, Moses Mosobo, Nelson Ouma, Nicole Achieng, Patience Kiyuka, Perpetual Wanjiku, Peter Mwaura, Rita Warui, Robinson Cheruiyot, Salim Mwarumba, Shaban Mwangi, Shadrack Mutua, Susan Njuguna, Victor Osoti, Wesley Cheruiyot, Wilfred Nyamu, Wilson Gumbi and Yiakon Sein
Data availability
Coastal Kenya genomes have been deposited to both GISAID (Accession numbers: EPI_ISL_1039223-29; EPI_ISL1440075-109, IPI_ISL_457845-931; EPI_ISL_568695-872; EPI_ISL806548-717; EPI_ISL_855494-548; EPI_ISL_968807-9245).
Funding
This work was supported by the National Institute for Health Research (NIHR) (project references 17/63/82 (PI Prof. James Nokes) and 16/136/33 (PI Prof. Mark Wooolhouse) using UK aid from the UK Government to support global health research, The UK Foreign, Commonwealth and Development Office and Wellcome Trust (grant# 220985). The views expressed in this publication are those of the author (s) and not necessarily those of NIHR, the Department of Health and Social Care, Foreign Commonwealth and Development Office, Wellcome Trust or the UK government. Some members of COVID-19 Testing Team at KWTRP were supported by funding received by Dr Marta Maia (BOHEMIA study funded UNITAID), Dr Francis Ndungu (Senior Fellowship and Research and Innovation Action (RIA) grants from EDCTP) and Prof. Anthony Scott (PCIVS grant from GAVI).
SUPPLEMENTAL MATERIAL

The SARS-CoV-2 epidemic in Kenya. Panel A, Map of Kenya showing county administrative boundaries and neighboring countries. The map is colored by the number of cases documented in the counties as at of 26th February 2021. Panel, B. Reported daily new cases in Kenya from March 2020 to February 2021 showing the first two national SARS-CoV-2 infection waves. Panel C, daily cases reported across coastal Kenya during the study period. Panel D, Kenya government intervention levels during the study period as measured by the Oxford stringency index.

Position of non-synonymous mutations in the 684 genomes from coastal Kenya compare with the Wuhan reference genome (Accession number NC_045512). Only changes occurring in >1% of genomes are shown. The percentage frequency of the changes in the 684 genomes is shown on the y-axis.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Panel A, root-to-tip regression plots for the four lineages first identified in Kenya. Panel B, Genome maps of the “Kenya” lineages where the spike region is shown in detail and in color and the rest of the genome is shown in grey color.
Acknowledgements
We thank (a) the members of the six Coastal counties of Kenya RRTs for collecting the samples analysed here; (b) the members of the COVID-19 KWTRP Testing Team who tirelessly analysed the samples received at KWTRP to identify positives (see full list of members below); (c) the KWTRP data entry team, (d) Laboratories that have shared sequence data on GISAID that we included as comparison data in our analysis (see list in appendix); (e) the KRISP team in South Africa for sharing the scripts we used in the import/export analysis and AFRICA-CDC for facilitating Africa genomics training. This paper is published with permission of the Director of KEMRI.
References



