
Abstract
Manuscript By lack of functional evidence, genome-based diagnostic rates cap at approximately 50% across diverse Mendelian diseases. Here we demonstrate the effectiveness of combining genomics, transcriptomics, and, for the first time, proteomics and phenotypic descriptors, in a systematic diagnostic approach to discover the genetic cause of mitochondrial diseases. On fibroblast cell lines from 145 individuals, tandem mass tag labelled proteomics detected approximately 8,000 proteins per sample and covered over 50% of all Mendelian disease-associated genes. By providing independent functional evidence, aberrant protein expression analysis allowed validation of candidate protein-destabilising variants and of variants leading to aberrant RNA expression. Overall, our integrative computational workflow led to genetic resolution for 21% of 121 genetically unsolved cases and to the discovery of two novel disease genes. With increasing democratization of high-throughput omics assays, our approach and code provide a blueprint for implementing multi-omics based Mendelian disease diagnostics in routine clinical practice.
The current ACMG recommendation for interpretation of genetic variants (Richards et al., 2015) attaches high importance to functional validation in designation of a variant as pathogenic or likely pathogenic. For this reason, systematic application of RNA sequencing (RNA-seq) has proven valuable in reducing the diagnostic shortfall of whole exome sequencing (WES) or whole genome sequencing (WGS) by providing a molecular diagnosis to 10% of unsolved cases with a mitochondrial disease (Kremer et al., 2017) and up to 35% in other disease cohorts (Cummings et al., 2017, Gonorazky et al., 2019, Fresard et al., 2019). However, while proteomics has been used to validate variants of uncertain significance (VUS) in single cases (Kremer et al., 2017, Lake et al., 2017, Borno et al., 2019, Stojanovski et al., 2020), the utility of systematic application into a diagnostic pipeline has yet to be explored.
Within the GENOMIT project (genomit.eu) we had analysed approximately 1,000 clinically suspected mitochondrial disease cases by WES/WGS and gathered corresponding Human Phenotype Ontology (HPO) terms for automated phenotype integration. Mitochondrial diseases are a prime example of the diagnostic challenge faced in human genetics given their vast clinical and genetic heterogeneity. In-keeping with previous studies (Stenton and Prokisch 2020), we reached a diagnosis by WES/WGS analysis for approximately 50% of the cases. Here, selecting 143 of these mitochondrial disease cases (121 unsolved and 22 solved positive controls) plus two healthy controls with available fibroblast cell lines, we performed RNA-seq and quantitative tandem mass tag (TMT) labelled proteomics in an integrative multi-omic approach (Fig.1a, Supplementary Fig. 1a-c) (see online Methods).

a, Multi-omic approach based on the integration of genomics (WES/WGS), transcriptomics (RNA-seq), proteomics, and phenotypic descriptors (HPO). We obtained DNA for WES/WGS from blood and RNA-seq and proteomics from fibroblasts obtained by minimally invasive skin biopsy. Functional evidence from each omic is integrated in search of a genetic diagnosis. The resultant diagnosis is thereby supported by multiple lines of robust clinical and functional evidence. Simultaneously, heterozygous and potentially biallelic genetic variants were prioritized according to their effect on the corresponding transcript(s) and protein by the identification of outliers in RNA-seq and proteomic data, in addition to aberrant splicing and monoallelic expression (MAE) of a deleterious heterozygous variants in RNA-seq data. Phenotype data complemented the analysis by gene-level prioritization based upon semantic similarity scoring. Together, omics integration allowed comprehensive gene-variant prioritization by providing insight into the effect of rare variation on expression of gene products. An overview of our multiplexed, time-efficient, RNA-seq and proteomic sample workflow is depicted in Supplementary Fig. 1a-c. b, Proportion of protein-coding genes detected by RNA-seq (blue) and proteomics (red), genome-wide and among mitochondrial disease genes (Schlieben and Prokisch 2020), Neuromuscular and Neurology genes (Frésard et al., 2019), and OMIM disease genes (https://omim.org). c, Protein z-score distribution for disease-causing genes in WES/WGS solved cases (positive controls). d, Protein z-score distribution for disease-causing genes with prioritized variants in WES/WGS unsolved cases. In panels c and d, the points appear in red for validated cases and in green for novel cases diagnosed in our downstream systematic approach.
With the detection of approximately 12,000 transcripts and 8,000 proteins per sample (Supplementary Fig. 1d), a median of 91% (n=353) and 80% (n=310) of mitochondrial disease gene products, and 59% (n=2535) and 51% (n=2159) of all Mendelian disease gene products were quantified per sample in RNA-seq and proteomics, respectively, deeming fibroblasts an easily accessible tissue with high disease gene coverage and an excellent resource for the study of mitochondrial diseases (Supplementary Fig. 1e).
To identify genes with aberrant RNA expression, we performed three outlier analyses, i) aberrant expression levels, ii) aberrant splicing, and iii) monoallelic expression of rare variants via the DROP pipeline (Yépez et al., 2021). To identify aberrant protein expression, we developed the algorithm PROTRIDER which estimates deviations from expected protein intensities while controlling for known and unknown sources of proteome-wide variation (see online Methods). In 18 positive controls with nuclear-encoded variants, detection of protein expression outliers in 14 (77.8%) validated our proteomic approach (Fig. 1c). In four positive controls with mtDNA-encoded variants, there was no significant change in protein expression resulting in a total validation rate of 64% across 22 positive controls (Supplementary Table 1).
In our cohort of 121 unsolved cases we first investigated those with variants prioritised in the WES/WGS analysis, spanning a total of 26 unique alleles across 21 cases, and mostly missense in nature (Supplementary Table 2). Variant pathogenicity was validated in 14 cases (67%) by nominally significant protein underexpression (Fig. 1d), of which five were also validated by aberrant RNA expression (Supplementary Fig. 1f). Moreover, proteomics was valuable in rejecting the prioritised variant, such as in the mitochondrial targeting sequence of MRPL53 which associated with normal expression of both MRPL53 and the large mitoribosomal subunit (Supplementary Fig. 1g).
Our matched genome, transcriptome, and proteome datasets together with protein expression outlier calls allowed us to investigate how aberrant RNA and protein expression relate to one another in the context of rare genetic variation, to our knowledge for the first time. After multiple-testing correction, we identified a median of two aberrantly expressed transcripts and six aberrantly expressed proteins per sample (Supplementary Fig. 2a). Though less than half of the RNA outliers resulted in significant protein outliers, possibly due to buffering mechanisms on the protein level, artefact, or lack of power, the majority (77%) did result in protein outliers (Fig 2a). The expression outliers were stratified into three classes: RNA-only, protein-only, and RNA-and-protein outliers. We focused on the two thirds of outliers that are underexpressed, as evidence for impaired function. All three classes of underexpression outliers were significantly enriched for rare variants in their encoding gene (Supplementary Fig. 2b-c). Within RNA-only outliers there was enrichment for splice, stop, and frameshift variants, in line with RNA expression outlier studies (Li et al., 2017). In contrast, protein-only outliers captured the functional consequence of missense variants and in-frame indels, demonstrating significant enrichment for coding variants. Substantially more RNA-and-protein outliers (15%) could be explained by potentially biallelic rare variants, compared to RNA-only and protein-only outliers (approximately 5%, respectively). An additional 25% of RNA-and- protein outliers were associated with rare heterozygous variants. Protein outliers without rare variants in the encoding gene may be explained indirectly as a consequence of protein complex instability due to a defect in one of the interaction partners (Kremer et al., 2017, Lake et al., 2017, Borno et al., 2019). Collectively, these genome-wide observations emphasize the complementarity of proteomics to RNA-seq in capturing the functional impact of rare genetic variation. Moreover it shows the sensitivity of the approach not only to biallelic variation, a hallmark of a recessive inheritance mode, but also to mono-allelic variation, i.e. those responsible for dominant diseases. If these biallelic or single variants are in-keeping with the inheritance mode and phenotype of the known disease gene, the detection of an outlier may lead to diagnosis of the patient.

a, RNA z-scores (y-axis) vs. protein z-scores (x-axis) detected by RNA-seq and proteomics across all samples. The shape indicates rare variants (minor allele frequency <1%) in the encoding gene. The size indicates semantic similarity with the established disease-gene associated phenotype. The colour represents outlier class. All detected splice defects and monoallelic expression (MAE) events resulted in aberrant expression, allowing this to be used as an indicator of variant pathogenicity. b, Individual OM06865 presented in childhood with predominantly neurological and muscular involvement. A homozygous missense variant in the autosomal recessive disease gene EPG5 was prioritised as a protein-only outlier. c, Individual OM27390 presented in infancy with failure to thrive, global developmental delay, seizures, encephalopathy, nystagmus, hypotonia, and abnormality of the basal ganglia on MRI. A heterozygous missense variant in the autosomal dominant disease gene MORC2 was prioritized by an RNA-and-protein outlier. g, Individual OM75740 presented in infancy with muscular hypotonia, cardiomyopathy, and abnormalities in the cerebral white matter on MRI. DARS2 was detected as an RNA-and-protein underexpression outlier explained by a combination of splice and intronic variants. h, Individual OM36526 presented in childhood with global developmental delay, elevated lactate, and an isolated respiratory chain complex (RCC) IV defect. The MRPS25 RNA-and-protein outlier is explained by a homozygous intronic variant demonstrating aberrant splicing. In addition, three other proteins from the small mitoribosomal subunit were underexpressed as a downstream consequence of the primary defect.
Aiming to pinpoint pathogenic genes and variants for those remaining cases without prioritized VUS from WES/WGS, we next combined aberrant expression analysis with patient phenotypic annotations (Fig. 2a, Supplementary Fig. 2d-e). Focussing on significant underexpression outliers (median 4 per sample), a median of one outlier matched with the patient phenotype as described by HPO annotations (see online Methods). Manual inspection and clinical interpretation of the outliers resulted in the diagnosis of 12 cases (11%) (Fig. 2b-e, Supplementary Fig. 3). In four cases, the identified protein-only outlier led to the diagnosis by providing functional validation of VUS in a gene not previously prioritised, yet in-keeping with the phenotype and mode of inheritance of the disease (Fig. 2b). In eight cases, we identified the diagnosis as a significant RNA-and-protein outlier. Of these eight cases, one case had a one exon deletion in NFU1 identified by follow-up WGS in compound heterozygosity with a missense variant resulting in 21% residual protein (z-score −7.8). One case has a heterozygous missense variant in MORC2 resulting in 69% residual protein (z-score −4.5) (Fig. 2c). Four cases demonstrated aberrant splicing resulting in protein outliers, such as a homozygous near splice variant in MRPL44 (z-score −5.5), deep intronic variants in TIMMDC1 (z-score −6.2) and MRPS25 (z-score −4.8), and in one case a direct splice variant on one allele and a unique combination of two frequent intronic variants on the second allele (allele frequency 7.2% and 21.8%, respectively) causing exon skipping in DARS2 (z-score −6.3) (Fig. 2d). In one case, compound heterozygous variants in VPS11 originally prioritised by WES were not validated given normal protein expression. However, underexpression of MRPS25 and five subunits of the small mitoribosomal subunit led to the diagnosis, exemplifying the added value of proteomics in detecting the consequence on all detected proteins and complexes (Fig. 2e). Finally, in two cases, our integrated omics approach led to the identification of novel mitochondrial disease genes, MRPL38 and LIG3. The MRPL38 outlier (z-score −5.8) (Fig. 3a) illuminated a pathogenic 5’UTR deletion (Fig. 3b). The functional relevance was confirmed by reduced abundance of the large mitoribosomal subunit (Fig. 3c) resulting in a severe reduction in mitochondrial translation rate rescued by the re-expression of wild-type MRPL38 (Fig. 3d). The LIG3 outlier (z-score −4.2) (Fig. 3e) reprioritised a heterozygous nonsense variant within the mitochondrial targeting sequence (Fig. 3f) affecting only the mitochondrial isoform in trans with a deep intronic variant causing aberrant splicing (Fig. 3f). As a dual localized nuclear and mitochondrial DNA ligase, a defect in LIG3 was expected to impact mitochondrial DNA replication, supported by mtDNA depletion and a combined OXPHOS defect in the muscle biopsy (Fig. 3g), significantly decreased protein levels of mtDNA encoded gene products (Fig. 3h), and impaired mtDNA repopulation (Fig. 3i). The downstream functional consequence of the LIG3 variants was reflected by 63 additional protein outliers. The four cases solved by protein-only outliers, included a hemizygous X-linked NDUFB11 missense variant resulting in aberrant protein underexpression (z-score −4.1) and pathologically low abundance of respiratory chain complex (RCC) I (50%) with no rare variants within any other RCCI subunit. The reduction in RCCI was most pronounced in the ND4-module to which NDUFB11 belongs (44% remaining, lowest in dataset), in-keeping with a second confirmed NDUFB11 case (55% remaining, second lowest in dataset). The detection of this variant in the unaffected grandfather indicated incomplete penetrance. Attributing pathogenicity to variants of incomplete penetrance, even in the presence of a phenotypic match, is an outstanding challenge in human genetics. However, in cases where reduced activity is causative of disease, proteomics has the power to classify variants affecting protein complex abundance. This was also demonstrated for a homozygous variant in DNAJC30 in two cases, by providing evidence for the loss-of-function character of an incompletely penetrant missense variant, as recently reported in a cohort of 27 families (Stenton et al., 2021).

a, Individual OM57837 presented in infancy with global developmental delay, intellectual disability, seizures, hypotonia, symmetrical basal ganglia and brainstem abnormalities on brain MRI, and respiratory chain complex (RCC) I and IV defects. The RNA and the protein products of MRPL38 (Mitochondrial Large Ribosomal Subunit Protein L38) were detected as underexpression outliers. b, A missense variant, c.[770C>G], p.[Pro257Arg], present in 78% of RNA reads indicated reduced expression of a compound heterozygous 127 bp deletion in the 5’UTR of MRPL38. c, Underexpression of MRPL38 resulted in reduction of the large mitoribosomal subunit (n=46 detected subunits). Meanwhile, the small mitoribosomal subunit remained unchanged (n=28 detected subunits). Data are displayed as a gene-wise protein expression volcano plot of nominal (-log10) p-values against protein intensity log2 fold change. d, Measurement of mitochondrial translation in cultured patient-derived fibroblasts by metabolic labelling with [35S]-containing amino acids. The MRPL38 mutant (P) showed a significantly reduced mitochondrial translation rate compared to control fibroblasts (C). e, Individual OM91786 presented with neonatal-onset severe encephalopathy, seizures, hypotonia, and increased serum lactate with early demise in the first weeks of life. LIG3 was identified as an RNA-and-protein outlier. f, Whole-genome sequencing identified compound heterozygous variants in LIG3. A nonsense variant within the mitochondrial targeting sequence affecting only the mitochondrial isoform of the protein in compound heterozygosity with a deep intronic variant leading to aberrant splicing and partial degradation of the transcript from this allele, indicated by allelic imbalance. g, A combined OXPHOS defect sparing nuclear encoded RCCII was present on the muscle biopsy. The black bars represent the reference range. The red point represents the patient measurement. h, Normal transcript level but reduced protein level of nuclear-encoded RCC subunits and ribosomal subunits (n=133) indicated complex instability. RNA-and-protein level of the 13 mtDNA encoded RNAs (11 mRNAs and 2 rRNAs) and 13 mtDNA encoded proteins were reduced. Expression is depicted as a mean-fold change compared to the mean of all other fibroblast samples. i, mtDNA copy number in cultured fibroblasts was investigated by qPCR during ethidium bromide induced depletion and repopulation. Impaired mtDNA repopulation was more severe than in the RNASEH1 mutant cell line which serves as a control for a repopulation defect (Reyes et al. 2015).
To summarise, leveraging on advanced proteomics we quantified a substantial fraction of expressed proteins, determined their normal physiological range in fibroblasts, and called protein outliers in a robust manner. By developing an integrated multi-omic analysis pipeline, we establish a clinical decision support tool for the diagnosis of Mendelian disorders. The power of proteomics is demonstrated by validation and detection of the molecular diagnosis in 26 of 121 (21%) unsolved WES/WGS cases, of which in 11 (42%) we detect downstream functional evidence on the complex level, explaining in total more than 100 outliers in these 26 cases (Fig. 4). Our code is freely available (https://prokischlab.github.io/omicsDiagnostics/). An interactive web interface allows the user to browse all results and could serve as a basis for developing future integrative multi-omics diagnostic interfaces. We used TMT-labelling, a proteomics technique quantifying the very same peptides for all samples of a batch. This greatly facilitates detection of under-expression outliers compared to conventional untargeted mass-spectrometry which suffers from widespread missing values in low intensity ranges. Though RNA-seq did not allow interpretation of missense variants, it provided independent cumulative evidence and guided the identification of causative splice variants in half of the solved cases. Moreover, RNA-seq has a deeper coverage of expressed genes, capturing 50% more genes. It therefore remains useful for lowly expressed proteins. To identify novel diagnoses we applied stringent significance filtering (FDR<0.1) and focussed on underexpression outliers with a phenotype match, leading to one protein outlier per sample in median. However, with the integration of multiple levels of omics information and phenotype descriptors, relaxed significance thresholds may in future be considered. Our approach depends on an available tissue, encouraging clinicians to be proactive and opportunistic in biosampling, specifically when follow-up visits are unlikely. Given the increasing democratization of proteomics we envisage its implementation in clinical practice to advance diagnostics by routine integration of functional data.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
A summary of the approach followed in the validation (left) and discovery (right) of protein outliers in our cohort of 121 unsolved mitochondrial disease cases to reach a molecular diagnosis in 26 cases (21%).
online Methods
Study cohort
All individuals included in the study or their legal guardians provided written informed consent before evaluation, in agreement with the Declaration of Helsinki and approved by the ethical committees of the centres participating in this study, where biological samples were obtained.
All studies were completed according to local approval of the ethical committee of the Technical University of Munich.
Cell culture
Primary fibroblast cell lines were cultured as per Kremer et al., 2017.
Whole exome sequencing (WES)
Whole exome sequencing was performed as per Kremer et al., 2017. SAMtools v.0.1.19 and GATK v.4.0 and called on the targeted exons and regions from the enrichment kit with a +/-50bp extension.
Variant annotation and handling
Variant Effect Predictor (McLaren et al., 2016) from Ensembl (Zerbino et al., 2018) was used to annotate genetic variants with minor allele frequencies from the 1000 Genomes Project (1000 Genome Consortium, 2015), gnomAD (Karczewski et al., 2020), and the UK Biobank (Bycroft et al., 2018), location, deleteriousness scores and predicted consequence with the highest impact among all possible transcripts. Variants with minor allele frequency less than 1% across all cohorts were considered as rare. Genes harbouring one rare allele were classified as rare, with two or more rare alleles - potentially biallelic. ACMG guidelines for variant classification were implemented with the InterVar software (Li and Wang, 2015).
Gene-phenotypic matching
Phenotype similarity was calculated as symmetric semantic similarity score with R::PCAN package (Godard and Page, 2016). We considered genes to match phenotypically if the symmetric semantic similarity between the gene and the case HPO annotations was larger or equal to 2 (Köhler et al., 2009; Frésard et al., 2019) (Supplementary Fig. 2d). Affected organ systems were visualized with R:: gganatogram (Maag 2018), based on patient’s HPO phenotypes corresponding to the third level of HPO ontology (Köhler et al., 2019).
RNA-sequencing
Non-strand specific RNA-seq was performed as per Kremer et al., 2017. Strand-specific RNA-seq was performed according to the TruSeq Stranded mRNA Sample Prep LS Protocol (Illumina, San Diego, CA, USA). Processing of RNA sequencing files was performed as per Kremer et al., 2017.
Detection of aberrant RNA expression, aberrant splicing, and mono-allelic expression
RNA-seq analysis was performed using DROP (Yepez et al., 2021), an integrative workflow that integrates quality controls, expression outlier calling with OUTRIDER (Brechtmann et al., 2018), splicing outlier calling with FRASER (Mertes et al., 2020), and mono-allelic expression with a negative binomial test (Kremer et al., 2017). We used as reference genome the GRCh37 primary assembly, release 29, of the GENCODE project (Frankish et al., 2019) which contains 60,829 genes. RNA expression outliers were defined as those with a false-discovery rate ≤ 0.1. Splicing outliers were defined as those with a gene-level false-discovery rate ≤ 0.1 and a deviation of the observed percent-spliced-in or splicing efficiency from their expected value larger than 0.3. Mono-allelic expression was assessed only for heterozygous single nucleotide variants reported by WES analysis. We retained mono-allelic expression calls at a false discovery rate ≤ 0.05. Aberrant events of all three types were further inspected using the Integrative Genome Viewer (Robinson et al., 2011).
Mass spectrometric sample preparation
Proteomics was performed at the BayBioMS core facility at the Technical University Munich, Freising, Germany. Fibroblast cell pellets containing 0.5 million cells were lysed under denaturing conditions in urea containing buffer and quantified using BCA Protein Assay Kit (Thermo Scientific). 15 µg of protein extract were further reduced, alkylated and the tryptic digest was performed using Trypsin Gold (Promega). Digests were acidified, desalted and TMT-labeling was performed according to (Zecha et al., 2019) using TMT 10-plex labelling reagent (Thermo Fisher Scientific). Each TMT-batch consisted of 8 patient samples and 2 reference samples common to all batches to allow for data normalization between batches. Each TMT 10-plex peptide mix was fractionated using trimodal mixed-mode chromatography as described (Yu et al., 2017). LC-MS measurements were conducted on a Fusion Lumos Tribrid mass spectrometer (Thermo Fisher Scientific) which was operated in data-dependent acquisition mode and multi-notch MS3 mode. Peptide identification was performed using MaxQuant version 1.6.3.4 (Tyanova et al., 2016) and protein groups obtained. Missing values were imputed with the minimal value across the dataset.
Transcriptome-proteome matching
In order to determine the correct assignment of proteome and transcriptome assay from the same sample, we correlated the gene counts with the protein intensities (Supplementary Fig. 4). The spearman ranked correlation test was applied to all transcriptome-proteome combinations, using the cor.test function from R. The distribution of the correlation values are plotted and in the case of mismatch two distinctive populations will appear. Correlations greater than 0.2 correspond to matching samples. Only protein intensities greater than 10,000 and genes with at least 50 counts were considered. Protein intensities were log-transformed and centered. RNA counts were normalized by sequencing depth using size factors (Love et al., 2014), log-transformed and centered. The 2,000 genes with the highest dispersion (as computed by OUTRIDER) were selected.
Detection of aberrant protein expression with PROTRIDER
To detect protein expression outliers while controlling for known and unknown sources of proteome-wide variations, we employed a denoising autoencoder based method, analogous to methods for calling RNA expression outliers (Brechtmann et al., 2018) and splicing outliers (Mertes et al., 2020). Specifically, sizefactor normalized and log-transformed protein intensities were centred protein-wise and used as input to a denoising autoencoder model with three layers (encoder, hidden space, decoder). As protein intensities varied strongly between batches, we included the batch as a covariate in the input of the encoder and in the input of the decoder (Supplementary Fig. 5a-b). For a given encoding dimension q, we fit the autoencoder by minimizing the mean squared error loss over the non-missing data. The optimal encoding dimension of the autoencoder was determined by artificially injecting outliers and selecting the encoding dimension that yielded the best area under the precision-recall curve (AUPRC) of recovering these injected outliers. For this dimension fitting procedure, artificial outliers were generated with a frequency of 1 per 1000. An outlier log-transformed intensity xoi,j for a sample i and a protein j was generated by shifting the observed log-transformed intensity xi,j by zi,j times the standard deviation σj of xi,j, with the absolute value of zi,j being drawn from a log-normal distribution with the mean of the logarithm equal to 3 and the standard deviation of the logarithm equal to 1.6, and with the sign of zi,j either up or down, drawn uniformly:
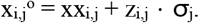 After the autoencoder model was fit to the data, statistical testing of the observed log-transformed intensities xi,j with respect to the expected log-transformed intensities μi,j modelled by the autoencoder was performed, using two-sided Gaussian p-values pi,j for sample i and protein j defined as
After the autoencoder model was fit to the data, statistical testing of the observed log-transformed intensities xi,j with respect to the expected log-transformed intensities μi,j modelled by the autoencoder was performed, using two-sided Gaussian p-values pi,j for sample i and protein j defined as
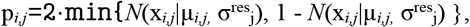 where σresj is the protein-wise standard deviation of the autoencoder residuals xi,j-µi,j. Finally, p-values were corrected for multiple testing per sample with the method of Benjamini and Yekutieli (Benjamini and Yekutieli, 2001). During the entire process of fitting the autoencoder model as well as the statistical tests, missing data was masked as unavailable and ignored. We refer to this method as PROTRIDER in the following.
where σresj is the protein-wise standard deviation of the autoencoder residuals xi,j-µi,j. Finally, p-values were corrected for multiple testing per sample with the method of Benjamini and Yekutieli (Benjamini and Yekutieli, 2001). During the entire process of fitting the autoencoder model as well as the statistical tests, missing data was masked as unavailable and ignored. We refer to this method as PROTRIDER in the following.
Benchmark of PROTRIDER against limma
As no method for outlier detection in proteomics data was established yet, we benchmarked our method against an approach that is based on limma (Smyth 2005), which was developed for differential expression analyses on microarray data and assesses statistical significance with a moderated t-statistic. We used recalibrated protein data which has been adjusted with respect to the two identical control samples in each MS-run as the input for limma and included the sex, batch and instrument annotation to adjust for confounding factors. To be able to use limma for outlier detection, we tested each sample against all other samples. We evaluated the performance of both methods based on precision-recall curves of detecting the known category I defects (Supplementary Fig. 5c-f). In this benchmark, PROTRIDER showed superior performance, as it was able to recover more known defects while reporting fewer outliers in total (median per sample of 6 vs. 8 for the limma based approach). Therefore, we decided to adopt PROTRIDER for the detection of aberrant protein expression.
Enrichment of genetic variants in outlier genes
We focused our analysis only on the genes where both RNA-and-protein levels were quantified, per every sample and limited it to the genes that were detected as outliers at least once in our cohort. Variants were stratified into six classes according to their impact on the protein sequence, defined by a combination of VEP (McLaren et al., 2016) annotations as follows: Stop (stop_lost, stop_gained), splice (splice_region_variant, splice_acceptor_variant, splice_donor_variant), frameshift (frameshift_variant), coding (missense_variant, protein_altering_variant, inframe_insertion, inframe_deletion), synonymous (synonymous_variant, stop_retained_variant) and non-coding (3_prime_UTR_variant, 5_prime_UTR_variant, downstream_gene_variant, upstream_gene_variant, intron_variant, non_coding_transcript_exon_variant, mature_miRNA_variant, intron_variant, intergenic_variant, regulatory_region_variant). Enrichment analysis was performed similarly as described by Li et al 2017, by modelling with logistic regression of each outlier category (RNA only, protein only, RNA-and-protein over- or underexpresssion) as a function of standardized variant class. For each gene, detected as an outlier of a particular category, the remaining set of individuals served as controls. Proportions of outlier genes were calculated by assignment of one variant class (out of six) with the highest significant enrichment in the corresponding outlier category.
Detection of aberrantly expressed protein complexes
Detection of aberrantly expressed protein complexes was performed similar to the differential protein complex expression method described by Zhou et al., 2019. Specifically, the quantified proteins were mapped to the protein complex database CORUM (v3.0) (Giurgiu et al., 2019) or to the mitochondria-related subset of HGNC gene groups by gene names. We considered the protein complexes of four subunits or more and with at least 50% of the subunits quantified. For each sample i and protein complex k, we computed yi,k, the mean deviation of observed versus expected protein intensities across all detected subunits (expressed in log2 fold-change and as estimated by PROTRIDER or LIMMA). For each protein complex k, we fitted by maximum likelihood a Gaussian on all yi,k with mean µk, and standard deviation k using the fitdistr function from the R package MASS (Venables and Ripley 2002). The two-sided Gaussian p-values for sample i and protein complex j was then computed as:
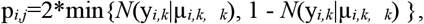 To correct the p-values for multiple testing, the method of Benjamini and Yekutieli (Benjamini and Yekutieli, 2001) was applied per every sample.
To correct the p-values for multiple testing, the method of Benjamini and Yekutieli (Benjamini and Yekutieli, 2001) was applied per every sample.
Mitochondrial translation assays
Metabolic labelling of mitochondrial proteins was performed essentially as described previously (Ruzzenente et al., 2018). In brief, fibroblasts derived from individuals #102875 and 98521 were incubated in methionine- and cysteine-free DMEM medium supplemented with 10% dialyzed FBS, GlutaMAX, sodium pyruvate (ThermoFisher Scientific, Montigny-le-Bretonneux, France), 100 mg/ml emetine dihydrochloride to block cytosolic protein synthesis and 400 μ Ci EasyTag EXPRESS35S Protein Labelling Mix (PerkinElmer, Villebon-sur-Yvette, France). Labelling was performed for 30 min followed by a further incubation for 10 min in standard growth medium. Equal amounts of total cell lysates were fractionated by SDS-PAGE and newly synthesized proteins were quantified by autoradiography.
Data and code availability
The proteomic raw data and MaxQuant search files have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository and can be accessed using the dataset identifier PXD022803. Code to reproduce the analysis is available via GitHub at github.com/prokischlab/omicsDiagnostics/.
Online resources
Code to reproduce the figures: https://github.com/prokischlab/omicsDiagnostics/tree/master
Web interfaces: https://prokischlab.github.io/omicsDiagnostics/#readme.html
PRIDE: https://www.ebi.ac.uk/pride/archive/projects/PXD022803
DROP: https://github.com/gagneurlab/drop
GTEx Portal: https://www.gtexportal.org/home/
OMIM database: www.omim.org
Author Contributions
Conceived and supervised the study, H.P; performed experiments, R.K, L.K, C.Lu, D.G, and M.M; analyzed and interpreted results, D.S, S.L, I.S, C.M, V.Y, D.G, M.M, R.K, S.L.S, J.G, H.P; provided essential materials, all authors; wrote the manuscript, S.L.S, H.P, J.G, R.K, and D.S; edited manuscript, all authors.
Competing Interests Statement
A.O. declares a consigned research fund (SBI Pharmaceuticals Co., Ltd.). All other authors declare no conflict of interest.
Acknowledgements
This study was supported by a German Federal Ministry of Education and Research (BMBF, Bonn, Germany) grant to the German Network for Mitochondrial Disorders (mitoNET, 01GM1906D), the German BMBF and Horizon2020 through the E-Rare project GENOMIT (01GM1920A), the ERA PerMed project PerMiM (01KU2016A), the German BMBF through the e:Med Networking fonds AbCD-Net (FKZ 01ZX1706A), the German Research Foundation/Deutsche Forschungsgemeinschaft (DI 1731/2-2), the AFM-Telethon grant (#19876), the Practical Research Project for Rare/Intractable Diseases from the Japan Agency for Medical Research and Development, AMED (JP19ek0109273, JP20ek0109468, JP20kk0305015, JP20ek0109485), a CMHI grant (S145/16), a PMU-FFF grant (A-20/01/040-WOS), the Pierfranco and Luisa Mariani Foundation (CM23), and the Italian Ministry of Health (GR-2016-02361494, GR-2016-02361241). We would like to thank Caterina Terrile, Franziska Hackbarth, and Hermine Kienberger for their excellent laboratory assistance as well as Miriam Abele for mass spectrometric support at the BayBioMS. We thank the “Cell line and DNA Bank of Genetic Movement Disorders and Mitochondrial Diseases” of the Telethon Network of Genetic Biobanks (grant GTB12001J) and Eurobiobank Network which supplied biological specimens.
References



