
Summary
Recent months have seen surges of SARS-CoV-2 infection across the globe along with considerable viral evolution1-3. Extensive mutations in the spike protein may threaten efficacy of vaccines and therapeutic monoclonal antibodies4. Two signature mutations of concern are E484K, which plays a crucial role in the loss of neutralizing activity of antibodies, and N501Y, a driver of rapid worldwide transmission of the B.1.1.7 lineage. Here, we report the emergence of a novel variant lineage B.1.526 that contains E484K and its alarming rise to dominance in New York City in recent months. This variant is partially or completely resistant to two therapeutic monoclonal antibodies in clinical use. It is also less susceptible to neutralization by convalescent plasma or vaccinee sera by 4.1-fold or 3.3-3.6-fold, respectively. The B.1.526 lineage has now been reported from at least 32 states in the US and numerous other countries. B.1526 has been outpacing B.1.1.7 in Northern Manhattan, and both variants have been spreading throughout New York with comparable estimated doubling times. Such transmission dynamics, together with its resistance to therapeutic antibodies, would warrant B.1.526 as a SARS-CoV-2 variant of concern.
Main
While evolution of SARS-CoV-2 was deemed to be slow at the beginning of the global pandemic5, at least four major variants of concern have emerged over the past four months1-3. These lineages are each characterized by numerous mutations in the spike protein, raising concerns that they may escape from therapeutic monoclonals and vaccine-induced antibodies. The hallmark mutation of B.1.1.7, the first described SARS-CoV-2 variant of concern that emerged in the UK, is N501Y located in the receptor-binding domain (RBD) of spike1. This variant is seemingly more transmissible and virulent6-8, perhaps due to a higher binding affinity of N501Y for ACE29. Two other variants of concern, B.1.351 (first detected in South Africa)2 and P.1 (first described in Brazilian travelers)3, share the N501Y mutation with B.1.1.7 but contain an E484K substitution in RBD2,3. Epidemiological evidence suggests that P.1 emerged as part of a second surge in Manaus, Brazil despite a high pre-existing seroprevalence of SARS-CoV-2 in the population. Reinfections with P.1, as well as with another related Brazilian variant P.2 that also harbors E484K, have been documented10,11.
Our previous study on B.1.351 demonstrated that this variant is refractory to neutralization by a number of monoclonal antibodies directed to the top of RBD, including several that have received emergency use authorization4. Moreover, this variant was markedly more resistant to neutralization by convalescent plasma and vaccinee sera. Importantly, these effects were in part mediated by the E484K mutation. These finding are worrisome in light of recent reports that three vaccine trials showed a substantial drop in efficacy in South Africa12-14. To systematically screen our patient population in Northern Manhattan for B.1.351 and other E484K variants such as P.1 and P.2, as well as B.1.1.7, we implemented a rapid PCR-based screen for signature mutations combined with genomic surveillance.
Rapid screening for signature SARS-CoV-2 mutations
We first developed rapid PCR-based single-nucleotide-polymorphism assays to search for N501Y and E484K mutations (see schematic in Extended Data Fig. 1) in clinical samples known to be positive for SARS-CoV-2 and stored in the Columbia University Biobank, a biorepository of SARS-CoV-2 patient specimens from hospitals and outpatient clinics within our medical system. Patient and clinical testing information was extracted from the COVID-Care database15. Between November 1, 2020 and March 5, 2021, a total of 38,987 nasopharyngeal swabs underwent clinical testing for SARS-CoV-2 at our medical center, with 3,350 positive samples identified; 2,353 positive samples were available through the Columbia University Biobank for our study. We screened 1,751 samples from this time-period (74.5% of all stored samples) for the two signature mutations. We identified 176 samples with E484K (13.0% of samples that showed a signal in our genotyping assay) and 41 (3.4%) with N501Y. Only one sample contained both mutations. The earliest case with E484K was collected in mid-November 2020. Subsequently, there was a substantial increase in E484K-positive cases over time (Fig. 1a), from 2.5% in early November to 8.9% by mid-January, and ultimately to 24.3% between February 21st and March 5th, 2021. Viruses harboring N501Y also increased over time, albeit more gradually, from the earliest detection in mid-January to 5.3% of screened isolates by the beginning of March.

(a) Detection of viruses with key signature mutations in spike over time. The earliest detected E484K-harboring variant was collected in mid-November 2020. The prevalence of E484K (nE484K/(nscreened + nE484K)) subsequently increased over time, from 4.8% in early December 2020 up to 24.3% in early March 2021. Throughout the study period, we identified fewer N501Y-than E484K-harboring isolates, with a maximum of 5.9% of N501Y during mid-February 2021. (b) Distribution of different viral lineages identified by whole genome sequencing. Within our collection (n=282), the majority of sequenced E484K and S477N fell within the B.1.526 lineage, and N501Y isolates were primarily but not exclusively within B.1.1.7. Other strains commonly identified in our hospital center include B.1.1.304, B.1.2, and a lineage L452R-harboring isolates within B.1. (c) Phylogenetic tree of SARS-CoV-2 variants identified by sequencing and alignment of key spike mutations. Unique patterns of spike protein mutations present in genomes sequenced from our hospital center with at least one mutation of interest or concern (E484K, N501Y, S477N, or L452R; n=64) are shown. Residues at which at least one sample harbored a mutation are displayed above the S-protein schematic.
Genomics of variant and wildtype SARS-CoV-2
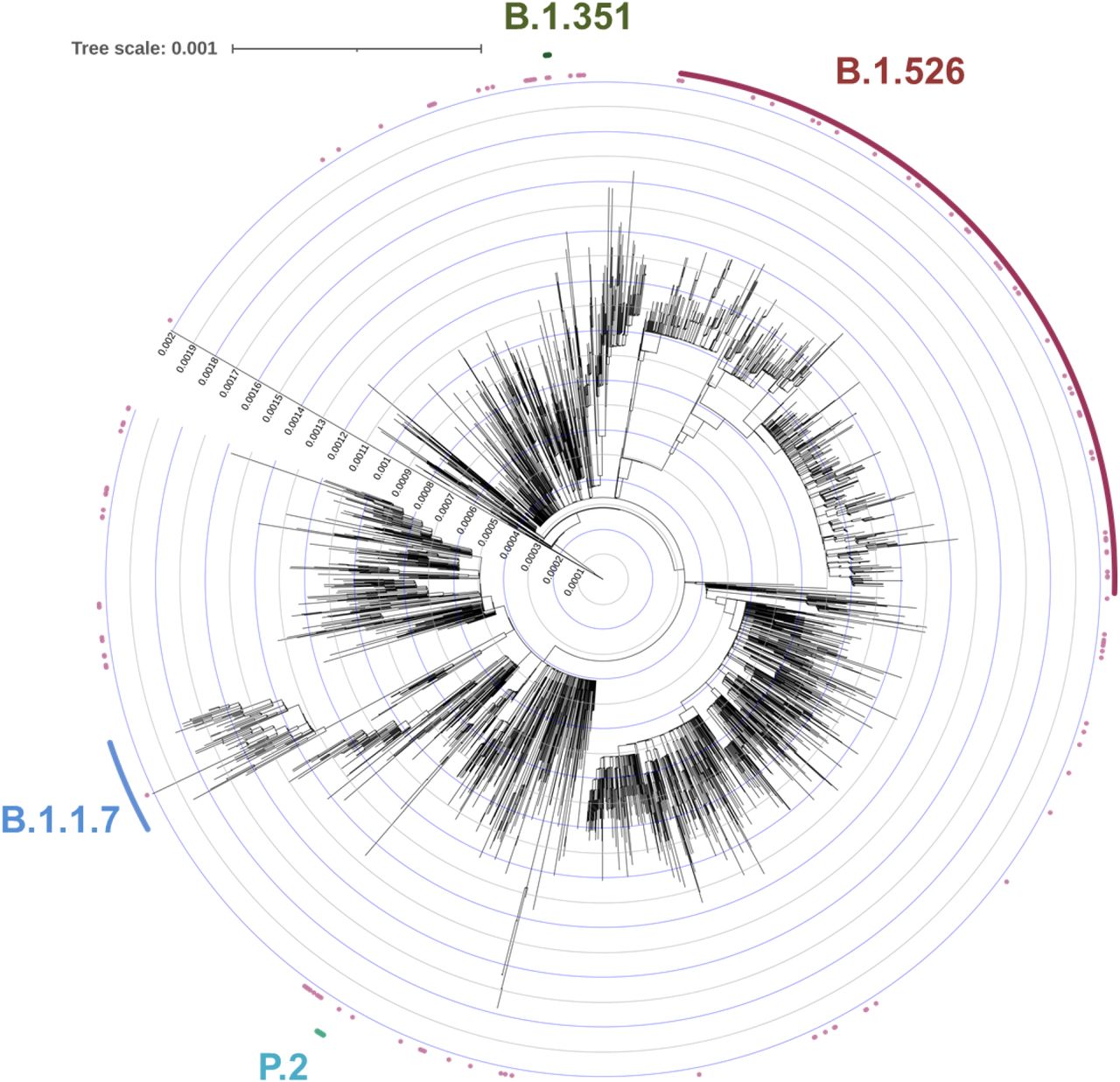
We performed whole genome nanopore sequencing on samples flagged as potential N501Y-or E484K-harboring strains with Ct values below 35 (n=132). We also sequenced samples negative for these signature mutations obtained during the same time period, all with Ct values below 35 (n=150). Sequencing results verified the E484K and N501Y substitutions in all samples identified by our screening PCR assays. Based on genomic sequencing, we performed phylogenetic analyses on all variant and non-variant genomes from our collection (Fig. 1b) and including publicly available genomic data (Extended Data Fig. 2). Amongst cases with N501Y, 17 (81.0% of 21 sequenced N501Y isolates) were identified as belonging to the B.1.1.7 lineage. One case with E484K was identified as P.2 and two as the parent lineage for the P.1 and P.2 strains, B.1.1.28. One sample which harbored both N501Y and E484K based on our screening assay was identified as B.1.351. However, quite unexpectedly, the large majority of the remaining cases with E484K (n=98/110, 89.1%) fell within a single lineage, B.1.52616. Further phylogenetic examination showed that the B.1.526 lineage is comprised of two sub-lineages harboring either E484K (B.1.526-E484K) or S477N (B.1.526-S477N) (Fig. 1c). From sequenced samples, which were negative for E484K and N501Y based on our PCR screen, we also noted the presence of B.1.526-S477N (n=26) in our collection. Strains that did not harbor E484K, N501Y, or S477N belonged to 29 distinct lineages; the most common were B.1.2 (n=28, 18.7%), B.1 (n=25, 16.7%), B.1.1.304 (n=12, 8.0%), and B.1.243 (n=11, 7.3%) among 150 sequences collected concurrently.
Signature mutations of the B.1.526 lineage
We first identified signature spike-protein mutations in the B.1.526 lineage by comparing all genomes generated as a part of this study. In Figure 1c, all unique patterns of S-gene mutations in our collection are displayed. Both B.1.526-E484K and B.1.526-S477N share characteristic spike-protein mutations L5F, T95I, D253G, D614G, and either A701V or Q957R along with either E484K or S477N. Non-spike mutations widely shared by B.1.526 isolates include: T85I in ORF1a-nsp2; L438P in ORF1a-nsp4, a 9bp deletion Δ106-108 in ORF1a-nsp6; P323L in ORF1b-nsp12; Q88H in ORF1b-nsp13; Q57H in ORF3a; and P199L and M234I in the N gene.
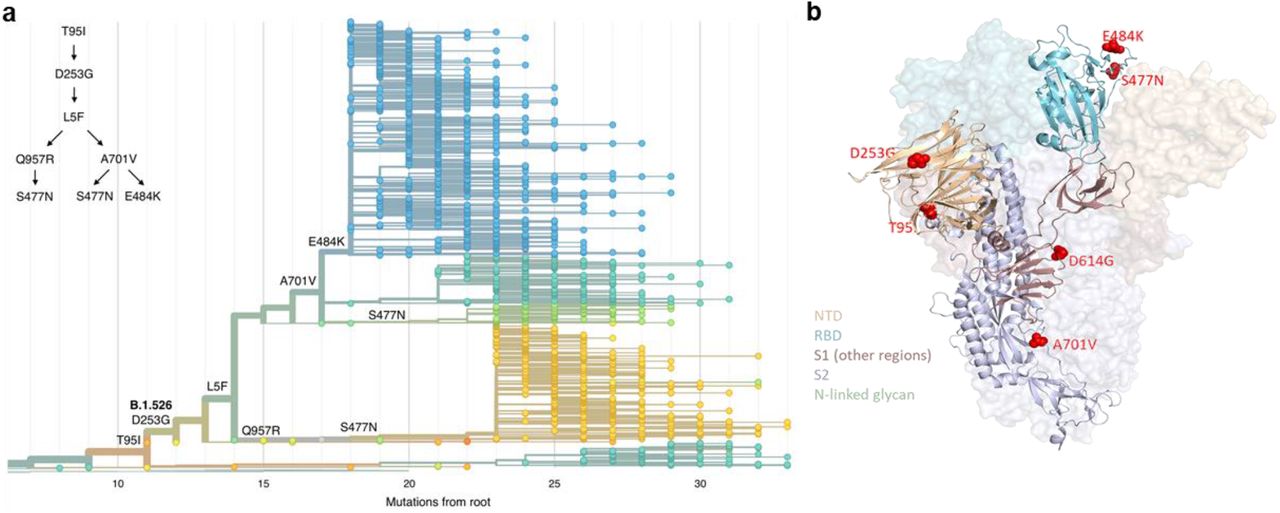
To further investigate the evolution of B.1.526, we performed phylogenetic analyses on genomes in this collection and in GISAID harboring the 9bp deletion Δ106-108 in ORF1a-nsp6, along with mutation A20262G that uniquely defines the parent clade containing B.1.526 and related viruses (Fig. 2a). We observed a stepwise emergence of these key mutations, with T95I, D253G, and L5F appearing in the earliest phylogenetic nodes. Isolates subsequently branched into three sub-lineages, with two major groups B.1.526-E484K and B.1.526-S477N containing A701V and one smaller sub-lineage B.1.526-S477N containing Q957R.
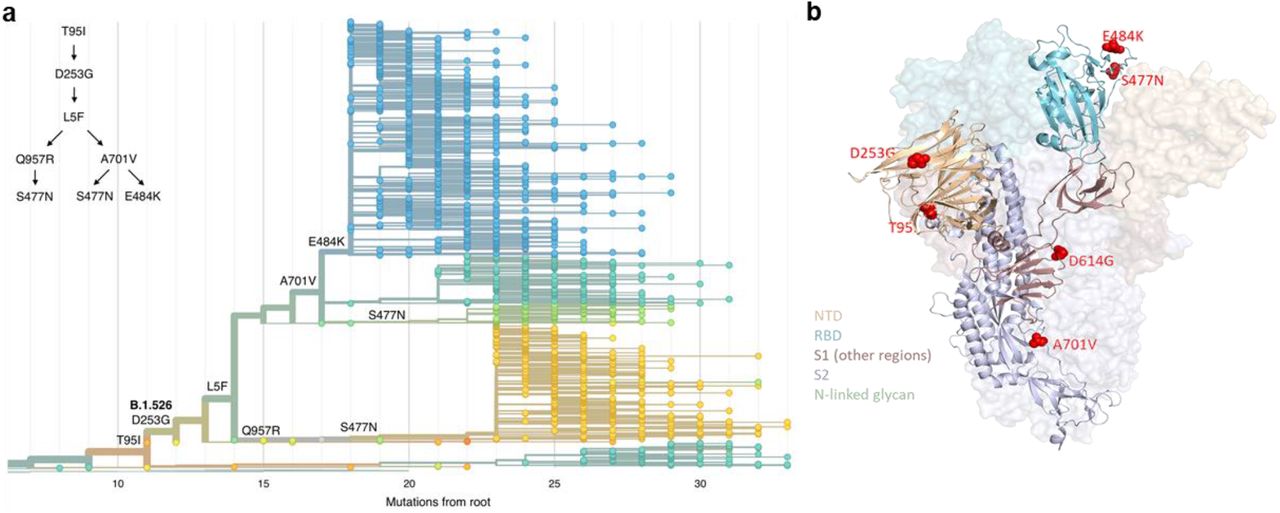
Three amino acid changes are characteristic of the B.1.526 lineage: L5F, T95I, D253G with sub-lineages possessing additional changes S477N, E484K, A701V and Q957R. (a) Maximum-likelihood phylogenetic tree of 2309 SARS-CoV-2 viruses colored according to spike protein haplotype. Spike protein mutations are labeled on the tree showing the stepwise accumulation of signature B.1.526 mutations and branching of the B.1.526-E484K and B.1.526-S477N sub-lineages. An interactive version of this figure is available at https://nextstrain.org/groups/blab/ncov/ny/B.1.526. (b) Key mutations of B.1.526 displayed on the spike trimer. The D253G mutation resides in the antigenic supersite within the N-terminal domain (NTD), a target for neutralizing antibodies, E484K and S477N at the receptor binding domain (RBD) interface with the cellular receptor ACE2, and A701V near the furin cleavage site.
Figure 2b displays the localization of B.1.526 signature spike mutations within the S protein structure. D253G resides in the antigenic supersite within the N-terminal domain17, which is a target for neutralizing antibodies18, whereas the E484K is situated at the RBD interface with the cellular receptor ACE2. The A701V mutation near the furin cleavage site is also shared with variant B.1.351.
Antibody neutralization of B.1.526
The impact of the signature S protein mutations in B.1.526 on antibody neutralization was assessed using vesicular stomatitis virus (VSV) based pseudoviruses as previously described4,18. Pseudoviruses containing S477N or E484K alone and all five signature mutations (L5F, T95I, D253G, A701V, and E484K or S477N), termed NYΔ5(E484K) or NYΔ5(S477N), were constructed and subjected to neutralization by 12 monoclonal antibodies including 4 with emergency use authorization, 20 convalescent plasma, and 22 vaccinee sera. The specifics of these monoclonal antibodies and clinical specimens were previously reported4. As shown in Figure 3a, the neutralizing activity of 12 monoclonal antibodies covering all epitopes on RBD was essentially unaltered against the S477N and NYΔ5(S477N) pseudoviruses, showing that this mutation has no discernible antigenic impact. However, against E484K and NYΔ5 pseudoviruses, the activities of several antibodies were either impaired or lost, including REGN10933 and LY-CoV555 that are already in clinical use. Likewise, neutralizing activities of convalescent plasma or vaccinee sera were lowered by 4.1-fold or 3.3-3.6-fold, respectively, against NYΔ5(E484K) (Fig. 3b), thereby raising the specter that risks of re-infection or vaccine breakthrough due to B.1.526-E484K may be heightened. A comparative analysis with other variants of concern (Fig. 3c) showed that such risks are likely lower than B.1.351 but higher than P.1 and B.1.1.7. Overall, these results demonstrate the need to modify our antibody therapy and to monitor the efficacy of current vaccines in regions where B.1.526-E484K is prevalent.

(a) Neutralizing activities of 12 monoclonal antibodies against pseudoviruses containing S477N or E484K alone and all five signature B.1.526 mutations (L5F, T95I, D253G, A701V, and E484K or S477N), termed NYΔ5(E484K) or NYΔ5(S477N). (b) Neutralizing activities of convalescent plasma (n=20) and vaccinee sera (n=22) against the NYΔ5(E484K) pseudovirus compared to wildtype pseudovirus. (c) Fold change in convalescent plasma and vaccinee sera neutralization ID50 of different variant pseudoviruses compared to wildtype pseudovirus. The data on B.1.1.7, B.1.351 and P.1 were derived from our prior publications4,23. Box and whisker plots show each measurement (symbol), along with median (center line), interquartile range (upper and lower bounds of each box), and the 9th and 91st percentile (whiskers) values.
Clinical comparisons of patients infected with E484K versus wildtype virus
Patients with E484K variant viruses were comparable in gender, age, race and ethnicity to those with wildtype SARS-CoV-2 (Extended Data Table 1). There were no significant differences in the rates of major comorbidities across E484K-positive and -negative groups, although patients harboring E484K isolates trended toward a higher rate of diabetes mellitus (30.8 vs 23.6%, p=0.07). The highest level of care required and maximal oxygen requirement were comparable between groups, although more patients with E484K isolates were still hospitalized at the time of this study (6.7 vs 2.8%, p=0.02). Notably, the cycle threshold (Ct) values for E484K isolates were significantly lower than wildtype isolates (mean 30.4 vs 31.8, p=0.01), indicating a modestly higher viral load in these variant samples.
Surge in B.1.526 across NYC and the U.S
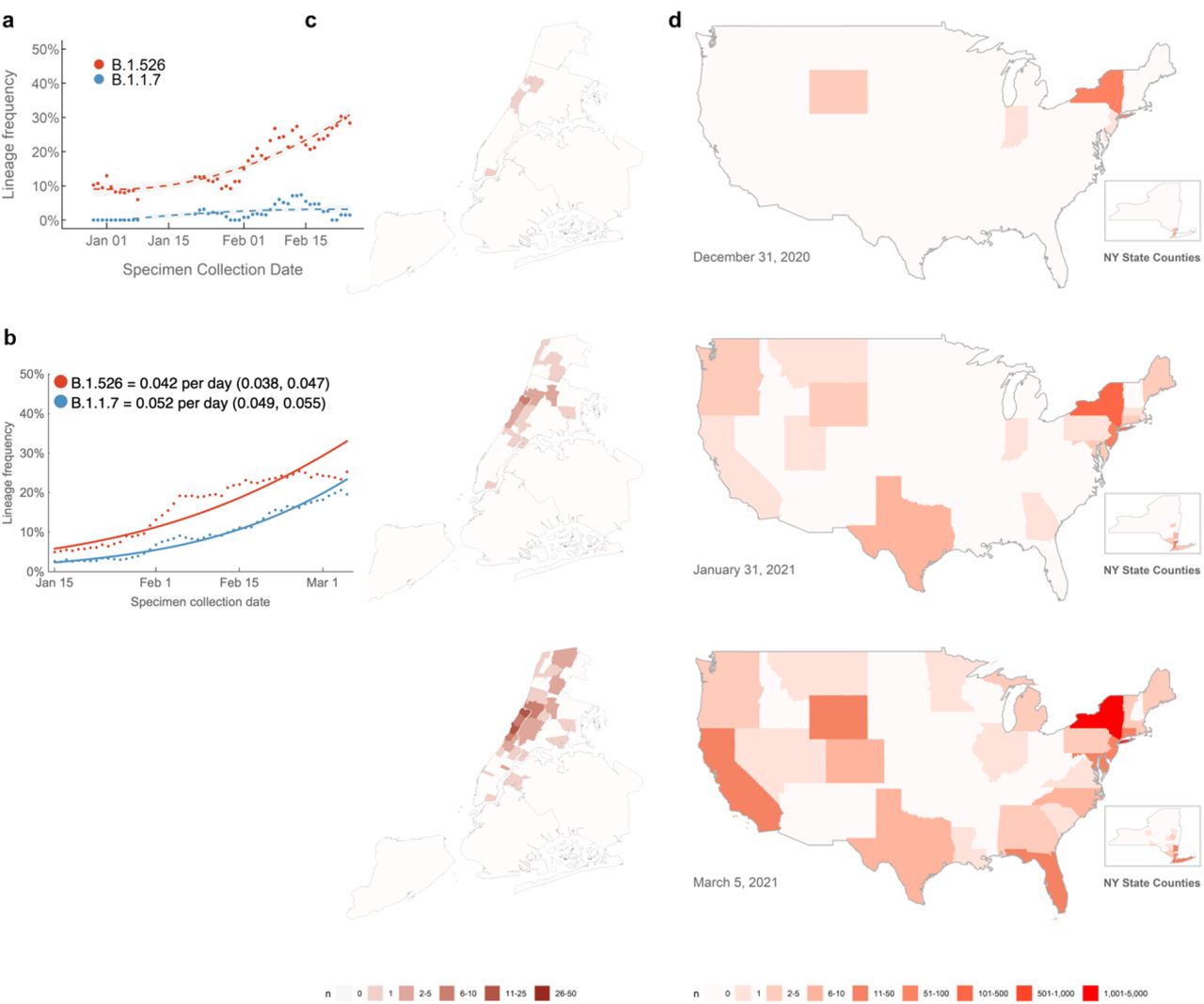
In recent weeks, we have observed a notable increase in isolates harboring not only B.1.526 with E484K, but also the separate sub-lineage with S477N (Fig. 1b). Prevalence of this novel variant B.1.526 has surged alarmingly in our hospital catchment area over the past few months, at a rate significantly outpacing that of B.1.1.7, with an estimated doubling time of 15 days (Fig. 4a). However, looking more broadly at publicly available data through March 21, we see similar logistic growth rates of B.1.1.7 and B.1.526 in viruses collected from New York State, with growth rate of B.1.526 estimated at 0.032 per day (95% CI 0.029–0.036) and growth rate of B.1.1.7 estimated at 0.040 per day (95% CI 0.037–0.043) (Fig. 4b). These differences may reflect distinct sampling and sequencing strategies. The data in Figure 4a represent unbiased comprehensive screening at a single site, which has consistently shown higher prevalence of B.1.526-E484K. In contrast, results in Figure 4b represent a larger geographic area and relies on public data subject to reporting biases. This may include preferential sequencing of samples with S-gene drop out in a diagnostic PCR8. At a minimum, B.1.526 is rising at a rate on par with B.1.1.7, a variant known to be substantially more transmissible19.
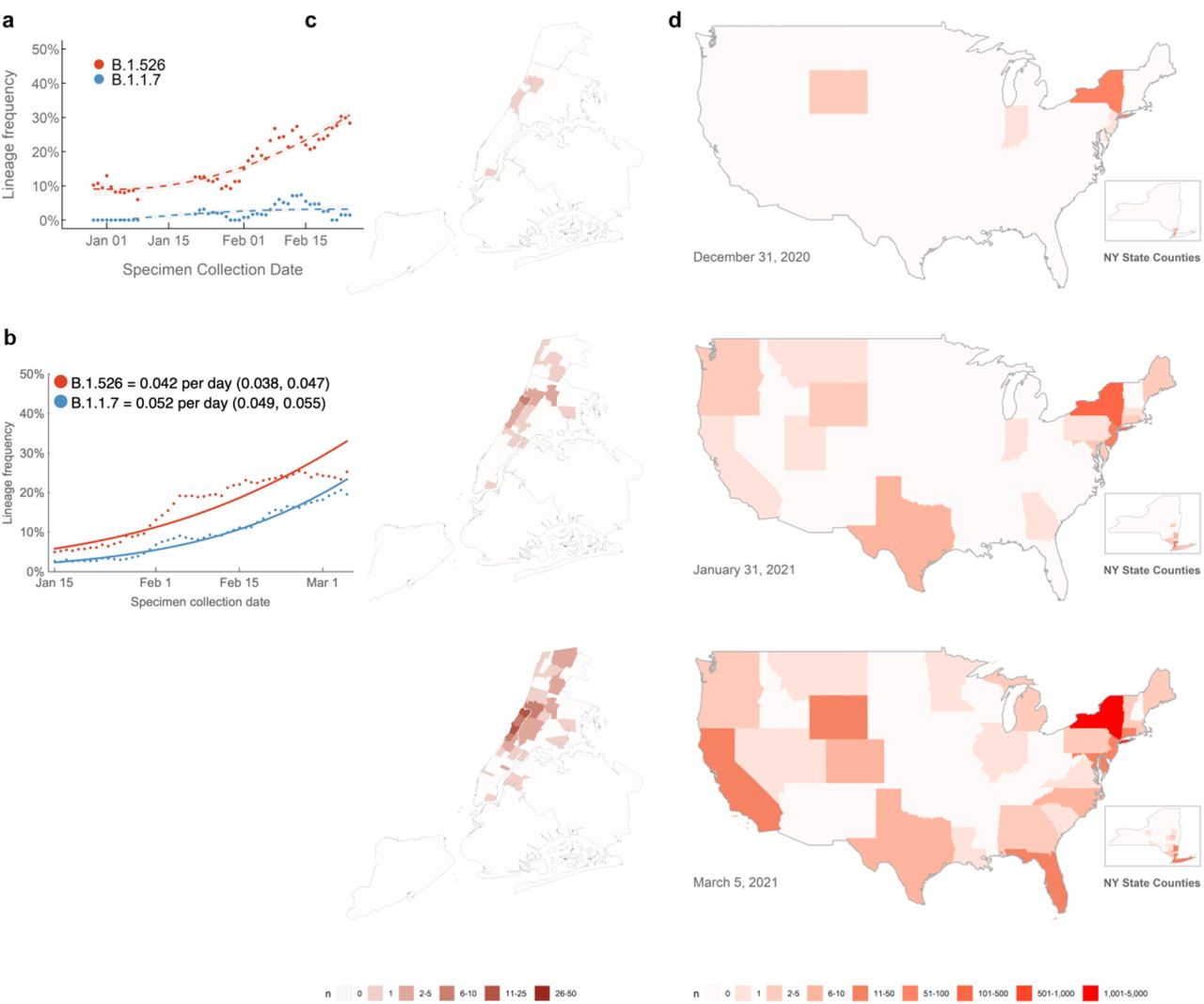
(a) Changing frequencies of B.1.526 versus B.1.1.7 in samples collected at Columbia University Irving Medical Center. Over the past four months, cases of B.1.526 have doubled more rapidly than B.1.1.7 in our hospital catchment area (every 15 days; 95% CI 3 – 22 days) and continue to rise at a steady rate. Dots show sliding 7-day average variant frequency and lines show logistic growth fit. (b) Changing frequencies of B.1.526 versus B.1.1.7 reported from New York State. Publicly available data from GISAID also shows consistent logistic growth of both B.1.526 and B.1.1.7 in sequences collected from New York State with growth rate of B.1.526 estimated at 0.032 per day (95% CI 0.029–0.036) and growth rate of B.1.1.7 estimated at 0.040 per day (95% CI 0.037–0.043). Dots show sliding 7-day average variant frequency and lines show logistic growth fit. (c) Increasing prevalence of B.1.526 in New York City. B.1.526 was initially concentrated in Upper Manhattan but has since spread throughout the city and beyond the immediate catchment area for our hospital center. (d) Spread of B.1.526 throughout the US. Early B.1.526 genomes from GISAID and from this study (n=61) were concentrated almost exclusively in New York. Genomes up to January 31, 2021 (n=396) show the beginning of B.1.526’s spread across the United States. As of March 5, 2021, 32 states have at least one confirmed case of B.1.526 (n=2,168), although the lineage is still most densely concentrated in New York. (Map inserts) Within New York, the spread of B.1.526 is evident from New York City to several counties in the Southeastern region of the state.
Patients with E484K variants were at first geographically concentrated in two distinct neighborhoods in the catchment area of our hospital system (Fig. 4c, top panel). However, especially by March 2021, many others were found scattered throughout the New York area without evidence for a single outbreak (Fig. 4c, middle and bottom panels). Indeed, based on data extracted from patient medical records, patients with SARS-CoV-2 harboring E484K were also more likely to reside inside rather than outside the greater metropolitan area, compared to those with non-variant strains (p=0.03, Extended Data Table 1).
Using patient location data for B.1.526 genomes generated as part of this study (n=132) as well as for sequences from GISAID (n=2,106) (see Supplementary Table 2)16, we found that B.1.526 genomes, primarily harboring E484K, were initially predominantly from samples collected in the Northeastern US (Fig. 4d, top panel). Even by March 2021 (Fig. 4d, middle and bottom panels), B.1.526 is concentrated in New York and surrounding states. This suggests that B.1.526, and B.1.526-E484K in particular, is now widespread in the region, the original epicenter of COVID-19 in the US20,21. Of increasing concern, however, is the spread of B.1.526 over the past two months to over 32 states across the country as well as internationally (Fig. 4d, middle and bottom panels).
Discussion
Here we report the emergence of E484K in a novel lineage, B.1.526, and its alarming surge in New York City. Neutralization assays using pseudoviruses containing mutations of the S protein in B.1.526 demonstrate that the activities of several antibodies were either impaired or lost, including two (Ly-CoV555 and REGN10933) already in clinical use (Fig. 3a). Furthermore, neutralizing activities of convalescent plasma or vaccinee sera were lower against B.1.526 harboring E484K (Fig. 3b). Going forward, it will be important to monitor the antigenic impact of this variant on re-infection or vaccine breakthrough. The S477N mutation, a key signature of another B.1.526 sub-lineage, did not have an impact on neutralizing monoclonal antibodies directed to the RBD (Fig. 3a). These findings underscore the importance of the E484K mutation, which has emerged in at least 108 different lineages of SARS-CoV-222, a real testament to convergent evolution. This raises the possibility that E484K can rapidly emerge in multiple clonal backgrounds and may warrant targeted screening for this key mutation in addition to robust genomic surveillance programs. The greatest threat of B.1.526 appears to be its ease of spread. It is keeping pace with B.1.1.7 in New York State (Fig. 4b) and surpassing it in the catchment area of our medical center (Fig. 4a). This apparent increased transmissibility is of great concern. Overall, the identification of B.1.526 reported here serves to highlight the need for concerted local, national, and international surveillance programs to track and contain the spread of novel SARS-CoV-2 variants.
Data Availability
All SARS-CoV-2 genomes generated as part of this study have been submitted to GISAID under submitter ID mka2136.
Methods
Clinical cohort
This observational study took place at an academic quaternary care center in New York City. Nasopharyngeal swabs obtained as part of routine clinical care were tested by the Clinical Microbiology laboratory, and positive specimens were transferred to the Columbia University Biobank for inactivation and storage.
Electronic health records data extracted for this analysis included demographics, laboratory results, admission, discharge, and transfer dates, current and historical international classification of disease (ICD 9 and 10) codes extracted from the clinical data warehouse. This study was reviewed and approved by the Columbia University Institutional Review Board (protocol number AAAT0123).
PCR screening
Extended Data Figure 1 describes our overall protocol for variant screening. To enable rapid PCR-based screening, we prepared RNA using the heat inactivation method in place of RNA isolation methods24. First, 50 µl of nasal swab sample in VTM solution was transferred into 96-well PCR plates, covered with an adhesive aluminum foil (VWR 60941-076) and incubated at 95°C for 5 min using the PCR instrument. After the centrifugation of the plate at >2,100 x g for 5 min, 5 µl of the supernatant from each sample, which contains viral RNA, was used for the SNP assay.
The SNP assay consists of four steps as follows: reverse transcription (RT) of viral RNA, pre-read of the SNP assay, real-time PCR and post-read of the SNP assay. 5 µl of RNA from the supernatant was added to 15 µl of the single step RT-qPCR reaction mix, which consists of 5 µl of TaqPath 1-step RT-qPCR Master Mix, CG (4x) (ThermoFisher Scientific), 500 nM of forward and reverse primers, 120 nM of VIC-MGB probe, 50 nM of FAM-MGB probe, 1/2000 volume of ROX Reference Dye (Invitrogen) as the final concentration, and nuclease-free water to adjust the total reaction volume of 20 µl. Each reaction plate included 8 control wells, 5×106 and 5×103 copies of WA-1 (wild type), UK variant and South African variant, which were generated by PCR to match the variant sequences, and 2 wells with water as no template controls (NTC).
The primer pairs and probes used are as follows. For the SNP assay for position 501, a primer pair of 501.F: 5’-GGT TTT AAT TGT TAC TTT CCT TTA CA-3’ and 501.R: 5’-AGT TCA AAA GAA AGT ACT ACT ACT CTG TAT G-3’ were used with two TaqMan probes (ThermoFisher Scientific), one for wild type, VIC.N501MGB: [VIC]-AA CCC ACT AAT GGT-MGBNFQ and the other for variant type, FAM.Y501MGB: [FAM]-AAC CCA CTT ATG GT-MGBNFQ. For position 484, a primer pair of 484.F: 5’-AGA GAG ATA TTT CAA CTG AAA TCT ATCAGG-3’and 484.R: 5’-GAA ACC ATA TGA TTG TAA AGG AAA GTA AC-3’ were used with two probes, one for wild type, VIC.E484MGB: [VIC]-ATG GTG TTG AAG GT-MGBNFQ and the other for variant type, FAM.K484MGB: [FAM]-ATG GTG TTA AAG GT-MGBNFQ. For position 477, the primer pair of 477.F and 477.R was used with two probes, one for wild type, VIC.S477MGB: [VIC]-TTA CAA GGT GTG CTA CCG-MGBNFQ and the other for variant type, FAM.N477MGB: [FAM]-TTA CAA GGT GTG TTA CCG-MGBNFQ.
The reaction plate was subjected to 1) reverse-transcription reaction (RT) at the condition at 25°C for 2 min, at 50°C for 15 min and a hold at 4°C; 2) SNP assay (pre-read) at 60°C for 30 sec; 3) real-time PCR at 95°C for 20 sec followed by 50 cycles of two-step PCR, at 95°C for 3 sec and at 60°C for 30 sec with the fast 7500 mode; followed by 4) SNP assay (post-read) at 60°C for 30 sec using ABI 7500 Fast Dx Real-Time PCR Instrument with SDS Software (ThermoFisher Scientific). The genotype at each key position for each sample was determined by reading the component signal of the amplification and the allelic discrimination analysis software in the program.
Whole genome sequencing
Isolates with cycle threshold (Ct) values below 35 were selected for sequencing using the ARTIC v3 low-cost protocol25. Briefly, RNA was extracted using the Qiagen RNeasy Mini kit or Zymo DNA/RNA Mini kit. Reverse transcription was performed using LunaScript RT SuperMix (NEB). Tiling PCR was performed on the cDNA, and amplicons were barcoded using the Oxford Nanopore Native Barcoding Expansion 96 kit. Pooled barcoded libraries were then sequenced on an Oxford Nanopore MinION sequencer using R9.4.1 flow cells. Basecalling was performed in the MinKNOW software v21.02.1. Sequencing runs were monitored in real-time using RAMPART (https://artic-network.github.io/rampart/) to ensure sufficient genomic coverage with minimal runtime. Consensus sequence generation was performed using the ARTIC bioinformatics pipeline (https://github.com/artic-network/artic-ncov2019). Genomes were manually curated by visually inspecting sequencing alignment files for verification of key residues in Geneious v10.2.6.
Phylogenetic analysis
For phylogenetic analysis of isolates sequenced at our hospital center (Fig. 1B-C), genomes were aligned using MAFFT v1.4.0 against the Wuhan-Hu-1 reference sequence (NC_045512.1). The resultant alignment was masked to avoid erroneous inclusion of SNPs due to sequencing errors as proposed by De Maio et al.26. To place our isolates in context with publicly available global sequencing data (Extended Data Fig. 2), we downloaded the Nextstrain North America dataset from GISAID which includes 4,029 genomes from 334 lineages, and all GISAID B.1.526 genomes collected between November 2020 and March 2021 (n=2,106). Public genomes were quality-filtered for <5% ambiguous bases and aligned with CUIMC genomes against the Wuhan-Hu-1 genome using MAFFT; duplicate sequences were removed, and the alignment was masked as above (see Supplementary Table 2 for acknowledgments of public genomic data utilized in this study). IQ-TREE v2.0.3 was used to generate phylogenetic reconstructions with 1,000 ultrafast bootstrap replicates. Interactive Tree of Life (iTOL) was used to visualize all phylogenetic tree figures.
Phylogenetic reconstruction of amino acid changes (Fig. 2A) was conducted using the Nextstrain27 workflow at https://github.com/nextstrain/ncov which aligns sequences against the Wuhan-Hu-1 reference via nextalign (https://github.com/nextstrain/nextclade), constructs a maximum-likelihood phylogenetic tree via IQ-TREE28, estimates molecular clock branch lengths via TreeTime29 and reconstructs nucleotide and amino acid changes also via TreeTime. This workflow was applied to 2309 SARS-CoV-2 genomes possessing the 9bp deletion Δ106-108 in ORF1a-nsp6 along with mutation A20262G which demarcates the parent clade to lineage B.1.526 alongside 688 global reference viruses. This analysis was conducted on data downloaded from gisaid.org30 on April 5, 2021.
Neutralization studies
We assayed the neutralizing activity of monoclonal antibodies (mAbs), convalescent plasma, and vaccinee sera against E484K, S477N, and WT (D614G) pseudoviruses, as well as pseudovirus NYΔ5 containing all five signature mutations of B.1.526-E484K (L5F, T95I, D253G, E484K, D614G, A701V), as previously described18. We examined four mAbs with emergency use authorization (CB6, REGN10987, REGN10933 and LY-CoV555), plus eight additional RBD mAbs, including ones from our own collection (2-15, 2-7, 1-57, & 2-36)18 as well as S30931, COV2-2196 & COV2-213032, and C12133, We also examined convalescent plasma collected in Spring of 2020 (n=20 patients), and Moderna and Pfizer vaccinee sera (n=22)4. Briefly, Vero E6 cells (ATCC) were seeded in 96-well plates (2 ×104 cells per well). Pseudoviruses were incubated with serial dilutions of the test samples in triplicate for 30 min at 37 °C. The mixture was added to cultured cells and incubated for an additional 24 h. Luminescence was measured using a Britelite plus Reporter Gene Assay System (PerkinElmer), and IC50 was defined as the dilution at which the relative light units were reduced by 50% compared with the virus control wells (virus + cells) after subtraction of the background in the control groups with cells only. The IC50 values were calculated using nonlinear regression in GraphPad Prism 8.0. Statistical analysis was performed using a Wilcoxon matched-pairs signed rank test. Two-tailed p-values are reported.
Growth dynamics
Growth rates of lineages B.1.1.7 and B.1.526 were estimated in Figure 4a using 7-day sliding window averages of the prevalence of B.1.1.7 and B.1.526, calculated as the number of sequence-verified samples from each strain divided by the total number of positive samples with cycle threshold (Ct) values below 35, as this threshold value was used for sequencing. For Figure 4b, prevalence data was obtained through by downloading “metadata” on April 5, 2021 for all 15,501 viruses from New York State collected after January 1, 2021. This metadata has PANGO lineages34 already assigned to each genome sequence. We calculated a 7-day sliding window of the frequency of B.1.1.7 and B.1.526 viruses in this dataset going from January 15 to March 21, 2021. This gives a timeseries of daily frequency estimates that we used to infer logistic growth rate by doing a logit transform on frequency followed by linear regression. The slope of a linear regression in logit space is equivalent to the growth rate of a logistic growth model following
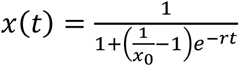 where x0 is the initial frequency and r is the logistic growth rate.
where x0 is the initial frequency and r is the logistic growth rate.
Data availability
All genomes and associated metadata generated as a part of this study have been uploaded to GISAID at the time of submission; accession numbers will be provided immediately once available and before final publication. Biological materials (i.e. variant pseudoviruses) generated as a part of this study will be made available but may require execution of a materials transfer agreement.
Code availability
Data processing and visualization was performed using publicly available software and packages, primarily RStudio v1.2.5033, GraphPad Prism v8.4, and iTOL (https://itol.embl.de/). The exact workflow used for phylogenetic analysis of public GISAID data (Fig. 2a) is available at https://github.com/blab/ncov-ny.
Competing Interests
P.W. and D.D.H. are inventors on a provisional patent application on monoclonal antibodies against SARS-CoV-2. D.D.H. is a member of the scientific advisory board of Brii Biosciences, which has provided a grant to Columbia University to support this and other studies on SARS-CoV-2. A.-C.U. has received funding from Merck & Co. unrelated to this study.
Author Contributions
Conceptualization – A.-C.U., D.D.H., M.K.A., H.M.; Data curation – M.K.A., H.M., J.E.Z., P.W., Z.S., T.B., A.G.-S., A.-C.U.; Formal analysis – M.K.A., P.W., J.E.Z., T.B., A.G.-S.; Funding acquisition – A.-C.U., D.D.H., M.K.A.; Investigation – M.K.A., H.M., J.E.Z., P.W., T.B.; Methodology – M.K.A., H.M., P.W., T.B.; Supervision – A.-C.U., D.D.H.; Visualization – M.K.A., P.W., T.B.; Writing – original draft – A.-C.U., M.K.A., H.M., D.D.H.; Writing – review and editing – all authors
Supplementary Information is available for this paper.
Correspondence and requests for materials should be addressed to Anne-Catrin Uhlemann (au2110{at}cumc.columbia.edu) or David D. Ho (dh2994{at}cumc.columbia.edu).
Extended Data
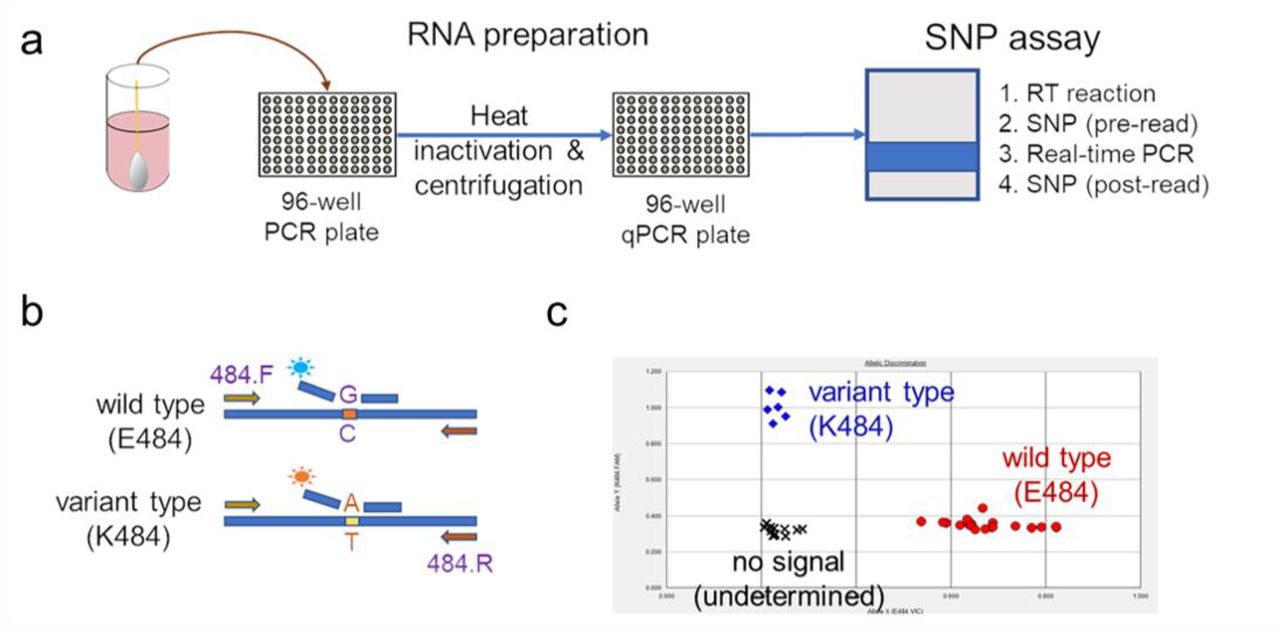
(a) Viral RNA is prepared by heat inactivation and centrifugation. The supernatant is then used for the SNP assay, which entails four steps: the reverse transcription (RT) reaction, pre-PCR reading of the plate to assess background fluorescence (SNP pre-read), real-time PCR, and post-PCR reading of the plate to measure fluorescence (SNP post-read). The runtime for this entire protocol is approximately two hours. (b) Genotype at targeted sites in COVID-19 viral RNA can be determined with two MGB probes, one for wild type (conjugated with VIC) and the other for variant type (conjugated with FAM). (c) Example signals for the variant type (K484; blue), the wild type (E484; red) and samples with no signal (black) are shown.

To place our B.1.526-E484K isolates within the context of globally circulating SARS-CoV-2 strains, we used the NextStrain North America dataset which includes genomes from 334 lineages, and all GISAID B.1.526 genomes collected between November 2020 and March 2021 (n=2,613 public genomes and 134 CUIMC genomes after quality-filtering and de-duplication). Isolates from this study assigned using Pangolin to B.1.1.7 (blue), B.1.351 (green), and P.2 (teal) fell within these branches, respectively. The majority of our isolates containing E484K fell within B.1.526 (dark pink).
Acknowledgements
Biospecimens utilized for this research were obtained from the Columbia University Biobank (CUB) with technical support from Viplan J. Mahadeva, Sebastian Fernando and Sylvia T. Parker-Jones. CUB is supported by the Irving Institute for Clinical and Translational Research (NCATS UL1TR001873). In particular, we thank Muredach Reilly, Eldad Hod, and the CUB COVID-19 Genomics Consortium (CCGC) for facilitating this effort. We are also grateful to Lihong Liu and Sho Iketani for technical support. We gratefully acknowledge all the authors, the originating laboratories responsible for obtaining the specimens, and the submitting laboratories for generating the genetic sequence and metadata and sharing via the GISAID Initiative, on which part of the presented research is based (see Supplementary Table 1). This work was in part funded by NIH/NIDA grant U01 DA053949 (A.-C.U, M.K.A.) and by support from Andrew & Peggy Cherng, Samuel Yin, Barbara Picower and the JBP Foundation, Brii Biosciences, Roger & David Wu, and the Bill and Melinda Gates Foundation. Funders and funding agencies had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
The manuscript was updated with additional sequencing data, new phylogenetic approaches and growth dynamic calculations, and additional pseudovirus antibody neutralization data.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}