
Abstract
The Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) was first detected in March 2020 in Uganda. Recently the epidemic showed a shift of SARS-CoV-2 variant distribution and we report here newly emerging A sub-lineages, A.23 and A.23.1, encoding replacements in the spike protein, nsp6, ORF8 and ORF9, with A.23.1 the major virus lineage now observed in Kampala. Although the clinical impact of the A.23.1 variant is not yet clear it is essential to continue careful monitoring of this variant, as well as rapid assessment of the consequences of the spike protein changes for vaccine efficacy.
Main Text
The novel Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2)(1) and the associated disease Coronavirus Disease 2019 (COVID-19)(2)(3) continue to spread throughout the world, causing >100 million infections and >2 million deaths (Johns Hopkins COVID-19 Dashboard). Genomic surveillance has played a key role in the responses to the pandemic; particularly, sequence data from SARS-CoV-2 provide information on the transmission patterns and the evolution of the virus as it enters new regions and spreads. As COVID-19 vaccines become available, monitoring SARS-CoV-2 genetic changes, especially changes at epitopes with implications for immune escape is crucial. A detailed classification system has been defined to help monitor SARS-CoV-2 as it evolves(4) and novel virus sequences first classified into 2 main phylogenetic lineages A and B representing the earliest divergence of SARS-CoV-2 in the pandemic and then into sub-lineages within these. Several Variants of Concern (VOCs) have emerged and spread with implications to compromise vaccine efficacy and/or therapeutic antibody treatments. These VOCs include lineage B.1.1.7 first identified in the UK (5), B.1.351 in South Africa (6) and lineage P.1 (B.1.1.28.1) in Brazil (7). The novel sub-lineage A (A.23.1) reported here encodes multiple spike, nsp6, ORF8 and ORF9 protein changes, and some of the replacements are predicted to be functionally similar to those observed in lineage B VOCs.
Status of the SARS-CoV-2 epidemic in Uganda
The first SARS-CoV-2 infection in Uganda was detected in March 2020. Initially the virus was detected among international travellers until passenger flights were stopped in late March 2020, a second route of virus entry was with truck drivers entering from adjacent countries (8). Since August 2020, community transmission has dominated the Uganda case numbers. By end of January 2021 detected case count in Uganda is 39,000, with 318 deaths attributed to SARS-CoV-2 infection. The SARS-CoV-2 infection has spread throughout the country, however the Kampala area is a major centre of virus infection where 60-80% of the daily new cases have been identified during the months of June 2020 to January 2021 (Uganda COVID-19 Daily Situation Report). We have continued our efforts to generate SARS-CoV-2 genomic sequence data to monitor virus movement and changes (8).
Changes in prevalence of lineage A viruses
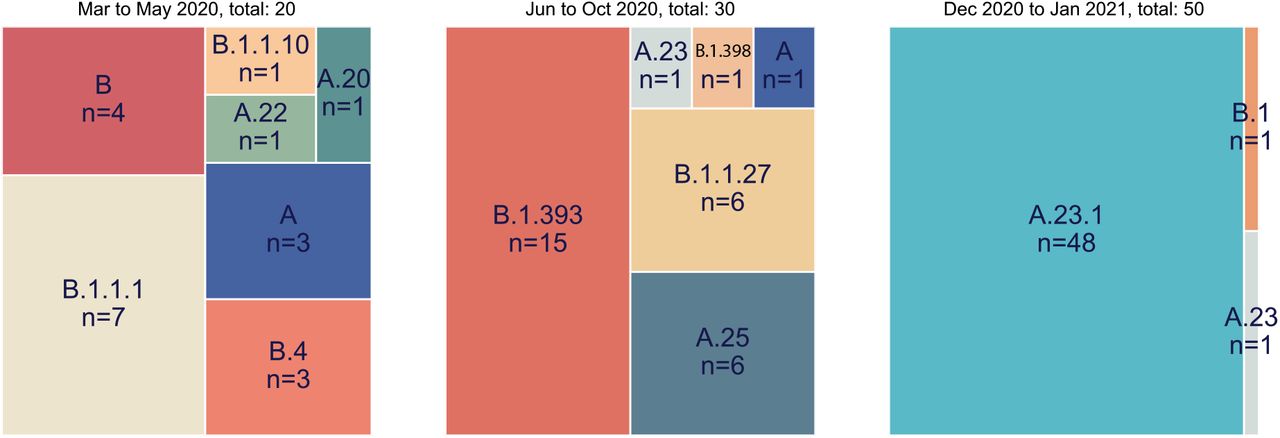
The genomes were classified into Pango lineages(4) using the Pangolin module pangoLEARN https://github.com/cov-lineages/pangolin) and into NextStrain clades using NextClade (9) (https://clades.nextstrain.org/). Across the entire epidemic, 80 (39%) strains belonged to the major lineage B whereas 123 (61%) strains were classified as lineage A (Supplementary Table 1). This distribution of lineages changed dramatically over the course of the year. A clear feature of the earlier COVID-19 epidemic in Uganda was the diversity of viruses found throughout the country attributed to frequent flights into Uganda from Europe, UK and Dubai (with origins further east); this is reflected in the range of lineages seen from March to May 2020 in the Kampala region (Figure 1, left panel). After passenger flights were limited in March, the virus was still able to enter via land travel, primarily with truck drivers. Uganda is landlocked country, characterised by its important geographical position, i.e. the crossing of two main routes of the Trans-Africa Highway in East Africa. The essential nature of produce and goods transport, therefore, resulted in potential virus movement from/to Kenya, South Sudan, DRC, Rwanda and Tanzania. In the period of June to October 2020 characterised by truck driver movement and no flights, lineage B.1 strains predominated, similar to pattern observed in Kenya(10) (Figure 1 middle panel). Given the diversity of virus lineages found in the country from March until October 2020, it was unexpected that by late December 2020 to January 2021 almost exclusively lineage A viruses (N=49) were found in Kampala with only one B.1 (Figure 1 right panel).In all time periods, the SARS-CoV-2 positive sample were obtained from 9 or more clinical locations throughout the Kampala region indicating that the differences are unlikely to be due to sampling different subpopulations in the city at different times.
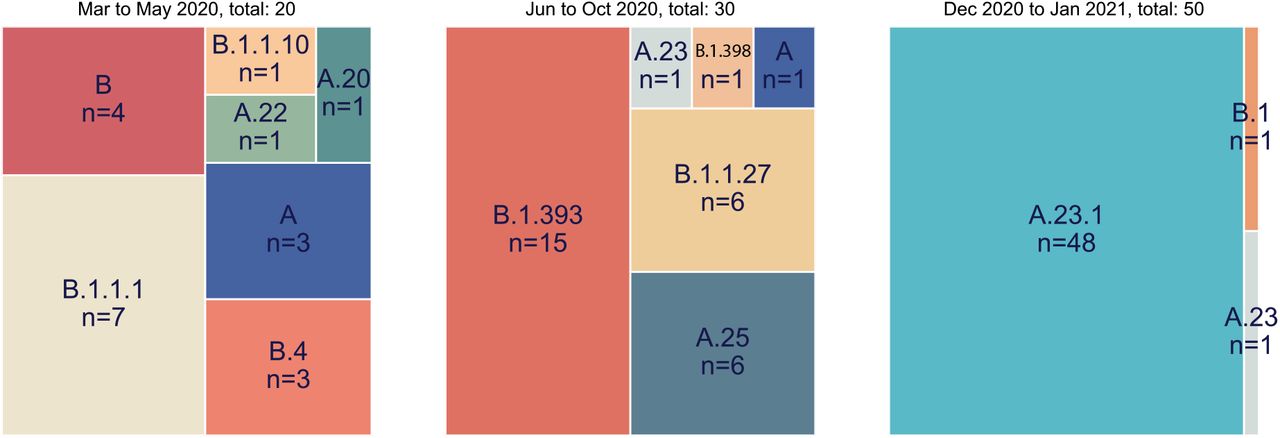
All full genomes from the Kampala area were lineage typed using the pangolin resource (https://github.com/cov-lineages/pangolin) Lineage counts were stratified into three collection periods (March-May 2020, June-October 202 and December 2020 to January 2021). The percentage of each lineage within each set was plotted as a treemap using squarify (https://github.com/laserson/squarify)with the size of each sector proportional to the number of genomes, genomes numbers are listed with “n=“.
Virus sequence diversity including fatal cases
To monitor the epidemic in more detail, full genome SARS-CoV-2 sequences were generated from SARS-CoV-2 positive samples in Uganda using 1500bp amplicon followed by MinION sequencing using modified 1500 bp amplicon method(11). All newly and previously generated genomes that are complete and high-coverage (N=203) were used to construct a maximum-likelihood phylogenetic tree (Figure 2). Important details to note: the genome sequences from 6 lethal Ugandan cases belonged to two lineages A.25 and Of note, the SARS-CoV-2 lineage A is far less prevalent than lineage B in Europe, UK and USA as compared to Asia. The presence of lineage A viruses from lethal and community cases throughout Uganda indicates that this lineage is circulating in Uganda and capable of producing a severe infection. Several variant lineages were observed at low frequencies and only briefly and may have undergone apparent extinction, similar to patterns observed in the UK (12) and Scotland (13).

Strain names are coloured according to the case profile (community: light brown, truck driver: dark green, return traveller: light green, lethal cases: red). The clusters from the Amuru (light blue), Kitgum (dark blue) and the recent community Kampala genomes are indicated. The tree was rooted where lineages A and B were split. The branch length is drawn to the scale of number of nucleotide substitutions per site, indicated in lower right.
A genome identified from a truck driver is often observed basal to each cluster (Figure 2), suggesting the importance of this route in the introduction and spread of the virus into Uganda. Additionally, genomes identified from truck drivers could provide important information, especially those truck drivers coming from ports of entries (POEs) bordering countries with limited genomic data on contemporary SARS-CoV-2 circulation, such as South Sudan and Tanzania (data not shown). As mentioned earlier, most of genomes identified from truck drivers coming from POEs bordering Kenya belonged to lineage B.1, consistent with the pattern reported in Kenyan cases (Supplementary Table 1). On the other hand, genomes identified from truck drivers from Tanzania, albeit small numbers, belonged to both A and B lineages (data not shown). From the small number of genomes from the Elegu POE bordering South Sudan, the viruses belonged to lineage A and B.1; the lineage A strain from this truck driver (strain UG053) was basal to the newly emergent A.23 variant (discussed below). Continued monitoring of all truck drivers coming in and out of the Uganda is therefore very important and would help us to better understand the inland cross-country import and export and circulation of strains in this part of world, where (large scale) genomic surveillance is scarce and resource is limited.
SARS-CoV-2 outbreaks in prisons
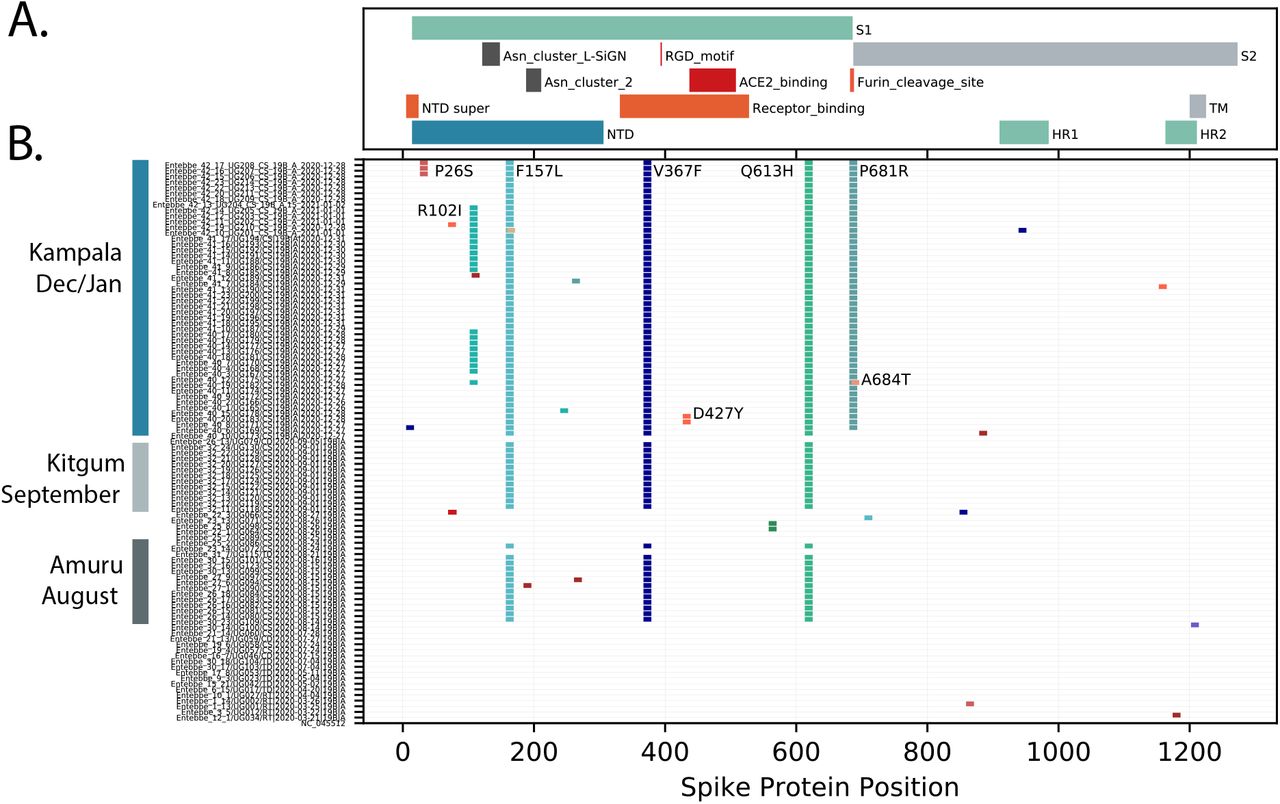
Outbreaks of SARS-CoV-2 infections were reported in the Amuru and Kitgum prisons in August 2020(14),(15). The SARS-CoV-2 genome sequences identified from individuals from Amuru and Kitgum prisons belonged exclusively to a lineage A (Figure 2) with three amino acid (aa) changes encoded in the spike protein (F157L, V367F and Q613H, Figure 3) that now define lineage A.23 (see below). Some individuals from Amuru prison were transferred to Kitgum prison, potentially facilitating virus movement between these prisons. By October 2020, lineage A.23 viruses were also found outside of the prisons in a community sample from Lira (a town 140 km from Amuru), and in two samples from the Kitgum hospital and in a community sample from Kampala. Lineage A viruses contributed to only 25% of the viruses in the Kampala region (Figure 1) in the period June to October 2020, which was consistent with the variety of virus entering Uganda and Kampala via international travellers and truck drivers seen in the initial period of the epidemic(8),(16). By December 2020, 49 of the 50 sequenced samples from the Kampala region (from 9 clinical sites) belonged to the new A.23.1 lineage (Figure 1 and 2) The pattern of mutations in these virus sequences was consistent with their evolution from the original A.23 viruses observed in Amuru/Kitgum cluster (Figure 2). A plot of nucleotide changes over time for Ugandan lineage A viruses (results not shown) showed a consistent evolutionary rate of roughly 2 nucleotide change per month that has been observed for SARS-CoV-2 throughout the pandemic (17),(18).

Lower panel: Each line represents the encoded spike protein sequence from a single genome, ordered by date of samples collection (bottom earliest, top most recent). Sequences from Amuru in August 2020, Kitgum in September 2020 and Kampala December 2020/January 2021 are indicated. Markers indicating the positions of amino acid (aa) differences from the reference strain, changes observed in multiple genomes are annotated with the annotation (original aa position new aa). Upper panel: The locations of important spike protein features are indicated. NTD: N-terminal domain, RBD: receptor-binding domain, S1: spike 1, S1: Spike 2, TM: transmembrane domain, HR1: helical repeat 1, HR2: helical repeat 2, NTD super: N-terminal domain supersite.
Important changes observed in the spike protein
The spike protein is crucial for virus entry into host cells, for tropism, and is a critical component of COVID-19 vaccine development and monitoring. The changes in spike protein observed in Uganda and global A.23 and A.23.1 viruses are shown in Figure 3. Many amino acid (aa) changes were single events with no apparent transmission observed. However, the initial lineage A.23 genomes from Amuru and Kitgum encoded three amino acid changes in the exposed S1 domain of spike, including F157L, V367F and Q613H (Figure 3). The V367F change is reported to modestly increase infectivity(19). Importantly, the Q613H change may have similar consequences as the D614G change observed in the B.1 lineage found predominantly in Europe and USA; in particular, D614G was reported to increase infectivity, spike trimer stability and furin cleavage (19),(20),(21),(22). These changes were not encoded by the closest known related genome (strain UG053) from a truck driver entering from South Sudan (Figure 2) and were not observed in previously reported genomes from Uganda (8).
Of concern, the recent Kampala and global A.23.1 virus sequences from December 2020-January 2021 now encoded 4 or 5 amino acid changes in the spike protein (now defining lineage A.23.1, see below) plus additional protein changes in nsp3, nsp6, ORF8 and ORF9 (Figure 3, 4). The P681R spike change encoded by all recent Kampala genomes since December 2020 adds a basic amino acid adjacent to the spike furin cleavage site. This same change has been shown in vitro to enhance the fusion activity of the SARS-CoV-2 spike protein, likely due to increased cleavage by the cellular furin protease (23); importantly, a similar change (P681H) is encoded by the recently emerging VOC B.1.1.7 that is now spreading globally across 75 countries as of 5 February 2021 (5) (24). There are also changes in the spike N-terminal domain (NTD), a known target of immune selection, observed in samples from Kampala A.23.1 lineage, including P26S and R102I plus 8 additional singleton changes (observed in only one genome, Figure 3).

All available SARS-CoV-2 complete genomes annotated as complete and lineage A from GISAID were retrieved on Feb 4 2021 and lineage typed using Pangolin(27). and confirmed as A.23 and A.23.1 by extracting examining the encoded spike protein. A.23 and A.23.1 genomes were plotted by country and sample collection date. Countries were anonymized by continent.
New lineage A designations
The viruses detected in Amuru and Kitgum met the criteria for a new SARS-CoV-2 lineage (25)(26) by clustering together on a global phylogenetic tree, sharing epidemiological history and source from a single geographical origin, and encoding multiple defining SNPs. These features including especially the three spike changes F157L, Q613H and V367F define the new lineage A.23. Continued circulation and evolution of A.23 in Uganda was observed and two additional changes in spike R102I and P681R were observed in December 2020 in Kampala; these SNPs define the sub-lineage A.23.1. Additional changes in non-spike regions also define the A.23 and A.23.1, including nsp3: E95K, nsp6: M86I, L98F, ORF 8: L84S, E92K and ORF9 N: S202N, Q418H. These new lineages can be assigned since pangolin version v2.1.10 and pangoLEARN data release 2021-02-01.
Screening SARS-CoV-2 genomic data from GISAID, viruses from A.23 and A.23.1 are now found in 12 countries outside Uganda (from Africa, Asia, Europe, North America and Oceania) indicating global movement of the newly emerging variants (Figure 4). In a screen of all available lineage A viruses in GISAID (Feb 4, 2021), the A.23 variant was first observed in Uganda in August 2020 and then in a country in North America (denotated as N.America_1) in October and in country in Africa (denotated as Africa_2) in December (Figure 4). The variant A.23.1 was first seen in December in 2020 although we have a 2-month gap in Uganda sequence data from October/November 2020. Outside of Uganda, A.23.1 was found in another country in Africa (Africa_3) from the end of November in 9 different countries across Europe (6 countries), Asia (2 countries) and Oceania (1 country). Of note, the international flights out of Uganda were restarted on 1 October 2020 with frequent flights to Europe and US overlaying via a country in Africa or Asia. Phylogenetic analysis supports the close evolution of A.23 to A.23.1 (Supplementary Figure 1).
Additional changes in Ugandan A.23 and A.23.1 genomes compared to other VOC genomes
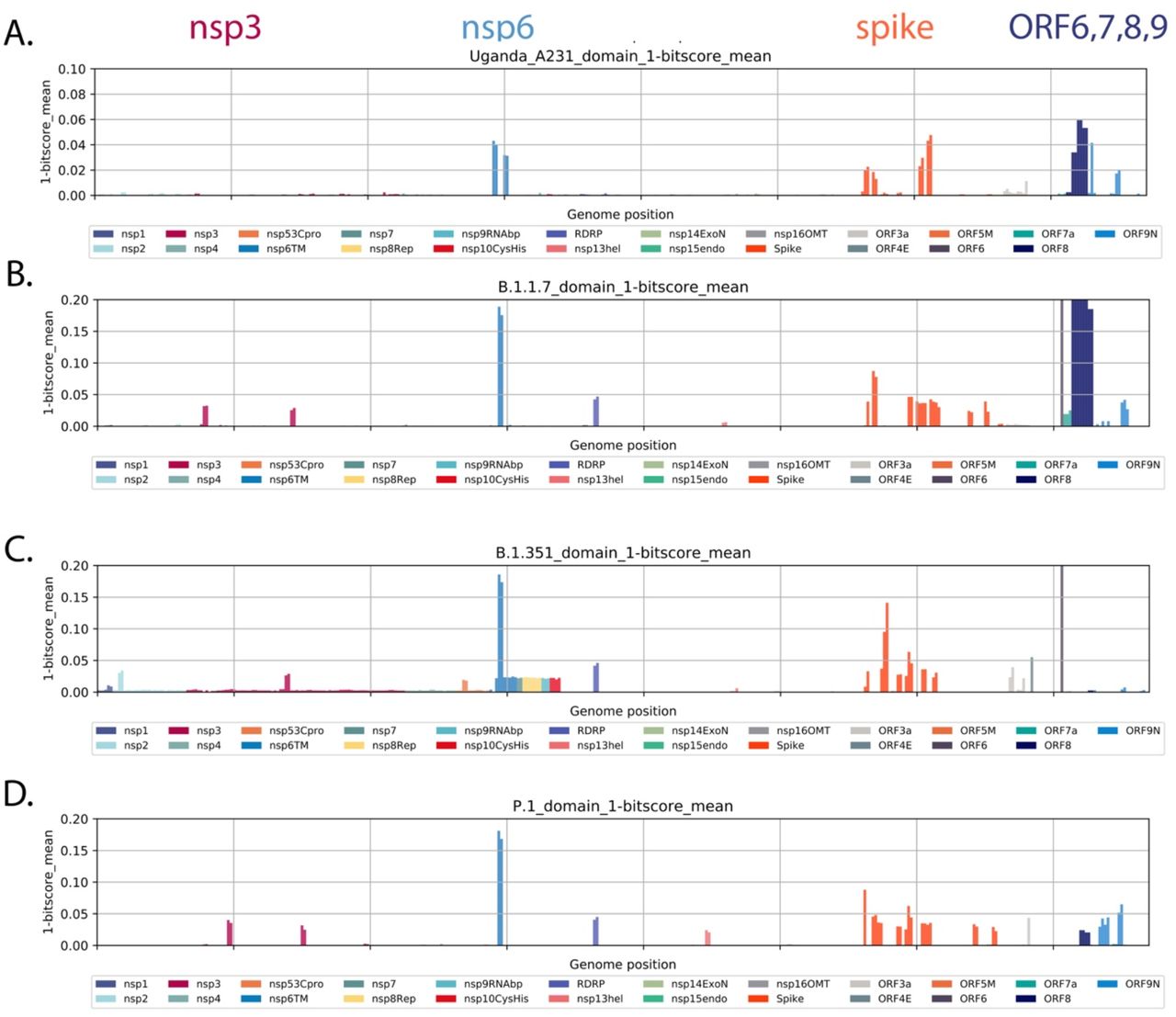
There are changes in other genomic regions of the virus accompanying the adaptation. We employed profile Hidden Markov Models (pHMMs) prepared from 44 amino acid peptides across the SARS-CoV-2 proteome to detect and visualize protein changes from the early lineage B reference strain NC_045512. Measuring the identity score (bit-scores) of each pHMMs across a query genome provides a measure of protein changes (in 44 amino acid steps) across the viral genome (Figure 5A). Applying this method to the most recent lineage A.23.1 genome sequences the changes in spike (discussed above) as well as changes in the transmembrane protein nsp6 and the interferon modulators ORF8 and 9 (Figure 5A). Modest changes were also observed in nsp13.

All forward open reading frames from the 35 early lineage B SARS-CoV-2 genomes were translated, and processed into 44 aa peptides (with 22 aa overlap), clustered at 0.65 identity using Uclust (28), aligned with MAAFT (29) and converted into pHMMs using HMMER-3(30). The presence of each domains and its bit-score (a measure of the similarity between the query sequence and the sequences used for the pHMM(30)) was sought in each set of SARS-CoV-2 VOC genomes and the 1-mean of the normalized domain bit-scores was plotted across the genome (e.g. 1 - the similarity of the identified query domain to the reference lineage B SARS-CoV-2 domain). Domains were coloured by the proteins from which they were derived with the colour code indicated below the figure. Panel A. Query set are 49 most Uganda lineage A.23.1 genomes. Panel B. All B.1.1.7 full genomes lacking Ns deposited in GISAID on Jan 26 2021, Panel C. All B.1.351 full genomes lacking Ns present in GISAID on Jan 26 2021, Panel D. All P.1 full genomes lacking Ns present in GISAID on Jan 26 2021.
We asked if a similar pattern of evolution was appearing in VOCs as SARS-CoV-2 adapted to human infection. We applied the same pHMM analysis to compare set of VOC or A.23.1 lineage viruses to the original SARS-CoV-2 lineage B genome sequences observed in December 2019/January 2020. The plots in Figure 5 show the difference in 44 amino acid steps across the viral proteome (see Methods for further details). We prepared sets of SARS-CoV-2 genomes annotated as B.1.1.7, B.1.351 or P.1 in GISAID and the A.23.1 genomes from this study. Both the A.23.1 and the B.1.1.7 lineage encode nsp6, spike, ORF8 and 9 changes (Figure 5B). Lineage B.1.351 encodes nsp6, spike and ORF6 changes (Figure 5C) and lineage P.1 encodes nsp6, spike and ORF6 changes (Figure 5D). Although the exact amino acid and positions of change within the proteins can differ in each lineage, there are some striking similarities in the common proteins that have been altered. Of interest, the nsp6 change present in B.1.1.7, B.1.351 and P.1 is a 3 amino acid deletion (106, 107 and 108) in a protein loop of nsp6 predicted to be on exterior of the autophagy vesicles on which the protein accumulates(31).The three amino acid nsp6 changes of lineage A.23.1 are L98F in the same exterior loop region, and the M86l and M183I changes predicted to be in intramembrane regions but adjacent to where the protein exits the membrane(31).
The spike changes affect the immunogenic N-terminal domain, the receptor binding domain, positions 613/614 (two lineages) and the furin cleavage site. A compilation of the amino acid changes in these lineages is found in Supplementary Table 2 with proteins that are altered in all 4 lineages marked in red.
Discussion
We report the emergence and spread of a new SARS-CoV-2 variant of the A lineage (A.23.1) with multiple protein changes throughout the viral genome. A similar phenomenon recently occurred with the B.1.1.7 lineage, detected first in the southeast of England (5) and now globally and with the B.1.351 lineage in South Africa(6), and P.1 lineage in Brazil(7) suggesting that local evolution and spread may be a common feature of SARS-CoV-2. Importantly, the lineage A.23.1 variant shares features found in the lineage B VOCs: alternation in key spike protein regions, especially the furin cleavage site and the 613/614 change that may increase spike multimer formation. The VOCs and A.23.1 strains also encode changes in similar region of the nsp6 protein which may be important for altering cellular autophagy pathways that promote replication. Changes or disruption of ORF7,8 and 9 are also present in the VOCs and A.23.1. The ORF8 changes or deletion probably indicates this protein is unnecessary for human replication, similar deletions accompanied SARS-CoV-2 adaption to humans(32),(33).
We suspect that all the emerging SARS-CoV-2 may be adjusting to infection and replication in humans and it is notable that the VOCs and lineage A.23.1 share a common patterns in their evolution. The spike changes are best understood due to the massive global effort to define the receptor and develop vaccines against the infection. The analysis reported in Figure 5 reveals common functions of SARS-CoV-2 that have been altered in all four variants, especially nsp6 and the ORF 7, 8 and 9. The functional consequences of the additional non-spike changes warrant additional studies and the current analysis may focus efforts of the proteins that are commonly changed in the variant lineages.
Methods
Sample collection, whole genome MinION sequencing and genome assembly
Residual nucleic acid extract from SARS-CoV-2 RT-PCR positive samples were obtained from Central Public Health Laboratory (Kampala, Uganda). The nucleic acid was converted to cDNA and amplified using SARS-CoV specific 1500bp-amplicon spanning the entire genome as previously described(11).The resulting DNA amplicons were used to prepare sequencing libraries, barcoded individually and then pooled to sequence on MinION R.9.4.1 flowcells, following the standard manufacturer’s protocol.
The genome assembies were performed as previously described (8). Briefly, reads from fast5 files were basecalled and demultiplexed using Guppy 3.6 running on the UMIC HPC. Adapters and primers sequences were removed using Porechop (https://github.com/rrwick/Porechop) and the resulting reads were mapped to the reference genome Wuhan-1 (GenBank NC_045512.2) using minimap2(34) and consensus genomes were generated in Geneious (Biomatters Ltd). Genome polishing was performed in Medaka, and SNPs and mismatches were checked and resolved by consulting raw reads.
Phylogenetic analyses
For the local Uganda virus comparison, all newly and previously generated genomes from Uganda (N=203, excluding 3 low coverage genomes) were aligned using MAFFT(29) and manually checked in AliView(35). The 5’ and 3’ untranslated regions (UTRs) were trimmed. Maximum-likelihood (ML) phylogenetic tree was constructed in IQTREE(36) under the GTR+F+R2 model as best-fitted substitution model according to Akaike Information Criterion (AIC) determined by ModelFinder in IQTREE and run for 100 pseudo-replicates. Resulting tree was visualised in Figtree(37) and rooted at the point of splitting lineage And B.
For phylogenetic analyses of Uganda lineage A.23 and A.23.1 strains comparing to global A.23 and A.23.1 strains, the global SARS-CoV-2 lineage A.23 and A.23.1 genomes were retrieved from GISAID on 4 February 2021 (N = 156). These global lineage A.23 and A.23.1 genomes combining with Ugandan A.23 and A.23.1 genomes (N = 97) and an outgroup of this A.23 lineage (Uganda strain UG053) were aligned using MAFFT and manually checked in AliView, followed by trimming 5’ and 3’ UTRs. From this alignment, the spike protein region was extracted for phylogenetic construction. The global and Ugandan A.23 and A.23.1 genomes were used to construct a ML tree under the GTR+F+I model as best-fitted substitution model according to AIC determined by ModelFinder and run for 100 pseudo-replicates in IQTREE. A ML tree was constructed in IQTREE for the nucleotide sequences encoding the spike protein from the global and Ugandan A.23 and A.23.1 strains, under the TPM2+F+I model of evolution as best-fitted model estimated by ModelFinder AIC and run for 100 pseudo-replicates. Resulting trees were visualised in Figtree and rooted using the strain UG053.
Ethical approvals
This study was approved by the Uganda Virus Research Institute-Research and Ethics Committee (UVRI-REC Federalwide Assurance [FWA] FWA No. 00001354, study reference. GC/127/20/04/771) and by the Uganda National Council for Science and Technology, reference number HS936ES. The novel reported SARS-CoV-2 genomes are available on GISAID (https://www.gisaid.org/) under the accession numbers EPI_ISL_954226-EPI_ISL_954300 and EPI_ISL_955136.
Author contribution statement
All authors contributed to the work presented in this paper.
Competing Interests statement
The authors declare no competing interests.
Supplementary Material


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
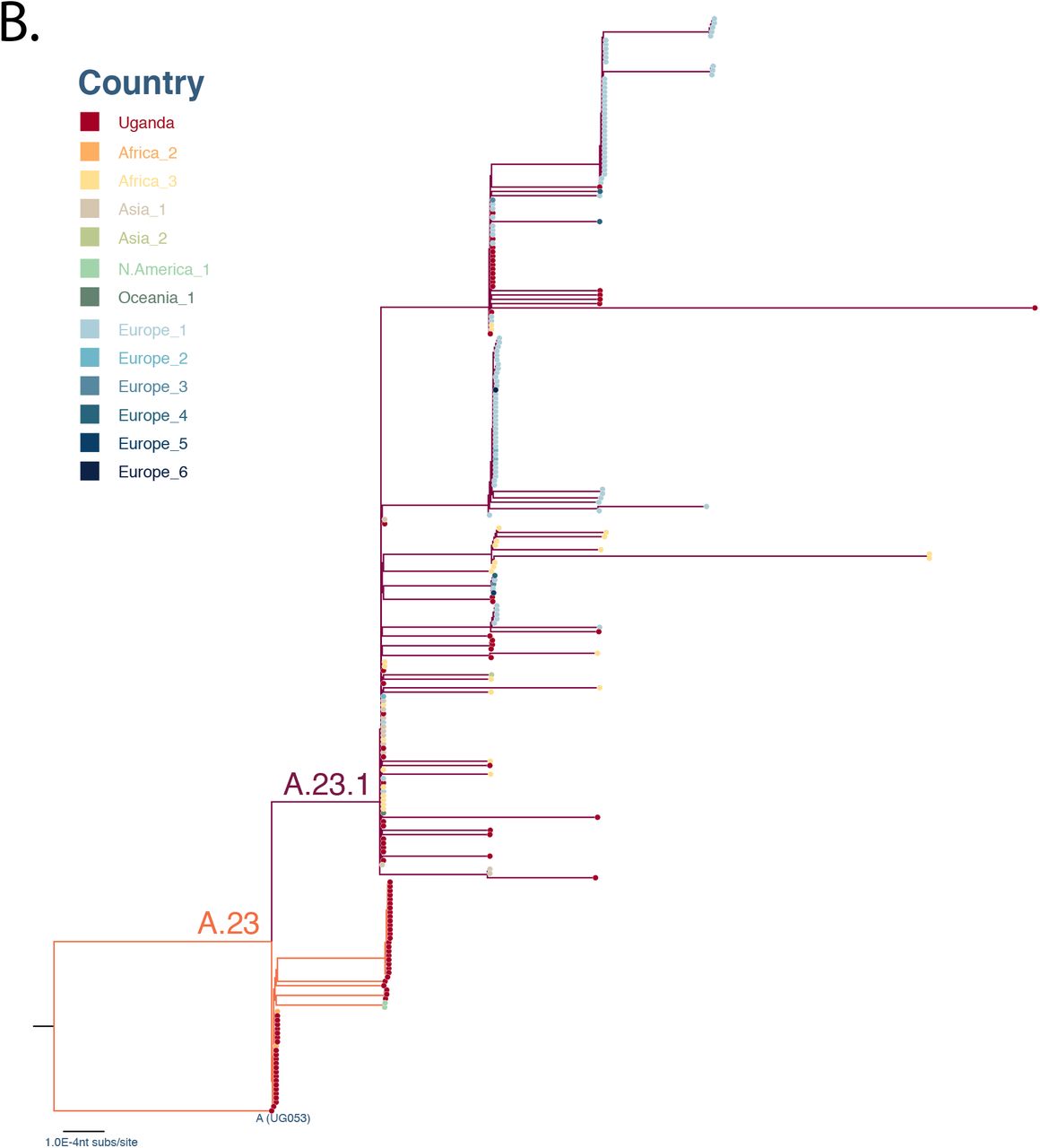
Panel A: A maximum-likelihood (ML) phylogenetic tree comparing Ugandan A.23 and A.23.1 (N = 97) with the global A.23 and A.23.1 (N = 156). Panel B: A ML phylogenetic tree of the nucleotide sequence encoding the spike protein region, comparing Ugandan A.23 and A.23.1 (N = 97) with the global A.23 and A.23.1 (N = 156). In both panels, the tree was rooted by the outgroup strain UG053 and strains were coloured according to the countries where they were identified. Branch length was drawn to the scale of number of nucleotide substitutions per site and trees were visualised in Figtree (37).
Lineage distribution in Uganda
Summary of replacements in the A.23.1 and 3 VOC lineages1.
Acknowledgements
We thank all global SARS-CoV-2 sequencing groups for their open and rapid sharing of sequence data and GISAID for providing an effective platform for making these data available. We are grateful to the Oxford Nanopore Technologies and the ARTIC Network for their support and we thank Pope Moseley for his constructive comments on the manuscript. The SARS-CoV2 diagnostic and sequencing award is jointly funded by the UK Medical Research Council (MRC) and the UK Department for International Development (DFID) under the MRC/DFID Concordat agreement (grant agreement number NC_PC_19060) and is also part of the EDCTP2 programme supported by the European Union. The UMIC high performance computer was supported by MRC (grant number MC_EX_MR/L016273/1) to PK. A.R. acknowledges the support of the Wellcome Trust (Collaborators Award 206298/Z/17/Z ARTIC network) and the European Research Council (grant agreement no. 725422 – ReservoirDOCS). The study is additionally funded by the Wellcome, DFID - Wellcome Epidemic Preparedness – Coronavirus (grant agreement number 220977/Z/20/Z) awarded to MC.
References



