
Abstract
SARS-CoV-2 transmission is uncontrolled in many parts of the world, compounded in some areas by higher transmission potential of the B1.1.7 variant now seen in 50 countries. It is unclear whether responses to SARS-CoV-2 vaccines based on the prototypic strain will be impacted by mutations found in B.1.1.7. Here we assessed immune responses following vaccination with mRNA-based vaccine BNT162b2. We measured neutralising antibody responses following a single immunization using pseudoviruses expressing the wild-type Spike protein or the 8 mutations found in the B.1.1.7 Spike protein. The vaccine sera exhibited a broad range of neutralizing titres against the wild-type pseudoviruses that were modestly reduced against B.1.1.7 variant. This reduction was also evident in sera from some convalescent patients. Decreased B.1.1.7 neutralization was also observed with monoclonal antibodies targeting the N-terminal domain (9 out of 10), the Receptor Binding Motif (RBM) (5 outof 29), but not in neutralizing mAbs binding outside the RBM. Introduction of the E484K mutation in a B.1.1.7 background to reflect newly emerging viruses in the UK led to a more substantial loss of neutralizing activity by vaccine-elicited antibodies over that conferred by the B.1.1.7 mutations alone. Further work is needed to establish the impact of these observations on protective vaccine efficacy in the context of the evolving B.1.1.7 lineage.
Introduction
The outbreak of a pneumonia of unknown cause in Wuhan, China in December 2019, culminated in a global pandemic due to a novel viral pathogen, now known to be SARS-COV-21. The unprecedented scientific response to this global challenge has led to the rapid development of vaccines aimed at preventing SARS-COV-2 infection and transmission. Continued viral evolution led to the emergence and selection of SARS-CoV-2 variants with enhanced infectivity/transmissibility2,3,4,5 and ability to circumvent drug6 and immune control7,8.
SARS-CoV-2 vaccines have recently been licensed that target the Spike (S) protein, either using mRNA or adenovirus vector technology with protection rates over a few months ranging from 62 to 95%9–11. The BNT162b2 vaccine encodes the full-length trimerised S protein of SARS CoV-2 and is formulated in lipid nanoparticles to optimise delivery to cells12. Other vaccines include the Moderna mRNA-1273 vaccine, which is also a lipid nanoparticle formulated S glycoprotein13 and the Oxford-AstraZeneca ChAdOx1 nCoV-19 vaccine (AZD1222) which is a replication-deficient chimpanzee adenoviral vector ChAdOx1, containing the S glycoprotein14. The duration of immunity conferred by these vaccines is as yet unknown. These vaccines were designed against the Wuhan-1 isolate discovered in 2019. Concerns have been raised as to whether these vaccines will be effective against new SARS-CoV-2 variants, such as B.1.1.7 (N501Y.V1), B.1.351 (N501Y.V2) and P1 (N501Y.V2) that originated in the UK, South Africa, and Brazil and are now being detected all over the world15–17.
The phase I/II studies of the Pfizer-BioNTech BNT162b2 vaccine determined the immunogenicity of different dosing regimens. The geometric mean concentration (GMC) of RBD-binding IgG 21 days after the first dose of 30 µg of the BNT162b2 vaccine, which is the dose approved in the UK, was higher than the GMC of a panel of convalescent plasma (1,536 vs 602 U/ml). Nevertheless, the corresponding neutralisation geometric mean titre (GMT) was 3-fold lower than a panel of convalescent plasma (29 vs 94)12, but substantially increased after boost immunization18. In older adults mean GMT was only 12 in a preliminary analysis of 12 participants19 and increased to 109 after the second dose.
In this study, we assess antibody responses induced three weeks after vaccination with the first dose of BNT162b2 following the rollout in the UK. In addition, by using a panel of human neutralizing monoclonal antibodies (mAbs) we show that the B.1.1.7 variant can escape neutralization mediated by most NTD-specific antibodies tested and by a fraction of RBM-specific antibodies. We also show that the recent appearance of the E484K mutation in B.1.1.7 isolates from the UK, similarly to the B.1.351 and P1 isolates, results in incremental loss of neutralization by BNT162b2 mRNA-elicited antibodies.
Results
Twenty-four participants had received the BNT162b2 mRNA vaccine three weeks prior to blood draw for serum and peripheral blood monocnulear cells (PBMC) collection. Median age was 82 years (IQR 64-85) and 30% were female (Supplementary Table 1). Serum IgG titres to N protein, S and the S RBD were assayed by particle based flow cytometry on a Luminex analyser (Fig. 1a). These data showed Spike and RBD antibody titres much higher than in healthy controls, similar to both convalescent plasma units used for therapeutic purposes as well as to serum from recovered individuals. The raised N titres relative to control could be the result of non specific cross reactivity that is increased following vaccination. However, the antibody response was heteorgenorus with almost 100-fold variation in IgG titres to S and Spike RBD across the vaccinated participants.
Vaccinee Participant ages

a, Serum IgG responses against N protein, Spike and the Spike Receptor Binding Domain (RBD) from 23 vaccinated participants (green), 20 recovered COVID-19 cases (red), 3 convalescent plasma units and 20 healthy controls (grey) as measured by a flow cytometry based Luminex assay. MFI, mean fluorescence intensity. b, Spike in open conformation with a single erect RBD (PDB: 6ZGG) in trimer axis vertical view with the locations of mutated residues highlighted in red spheres and labelled on the monomer with erect RBD. Vaccine (c-d) and convalescent sera (e-f) against WT and B.1.1.7 Spike mutant with N501Y, A570D, ΔH69/V70, Δ144/145, P681H, T716I, S982A and D1118H. g-h, neutralisation curves for serum from two convalescent individuals with reduced susceptibility to B.1.1.7 Spike mutant, means of technical replicates are plotted with error bars representing standard error of mean. Data are representative of 2 independent experiments.
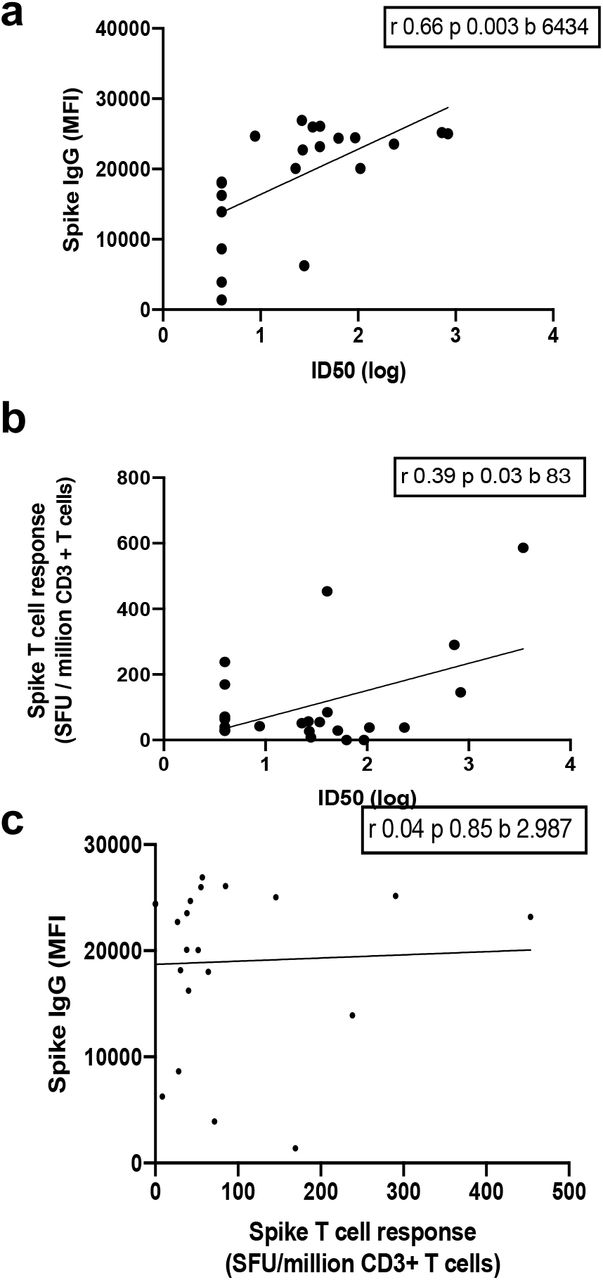
Using lentiviral pseudotyping we generated wild type S proteins on the surface of enveloped virions in order to measure neutralisation activity of vaccine-elicited sera. This system has been shown to give results correlating with replication competent authentic virus20,21. The vaccine sera exhibited a range of inhibitory dilutions giving 50% neutralisation (ID50) (Fig. 1c-d). The GMT against wild type (WT) pseudovirus was 24. Seven out of 23 participants exhibited no appreciable neutralisation against the WT pseudotyped virus. There was reasonable correlation between full length S IgG titres and serum neutralisation titres (Extended Data Fig. 1a). A broad range of T cell responses was measured by IFN gamma FLUOROSPOT against SARS-CoV-2 peptides in vaccinees. These cell responses did not correlate with IgG S antibody titres, but there was some correlation with serum neutralisation against WT virus (Extended Data Fig. 1b-c).

a, Relationship between serum IgG responses as measured by flow cytometry and serum neutralisation ID50. b, Relationship between serum neutralisation ID50 and T cell responses against SARS-CoV-2 by IFN gamma ELISPOT. SFU: spot forming units. c, Relationship between serum IgG responses and T cell responses.
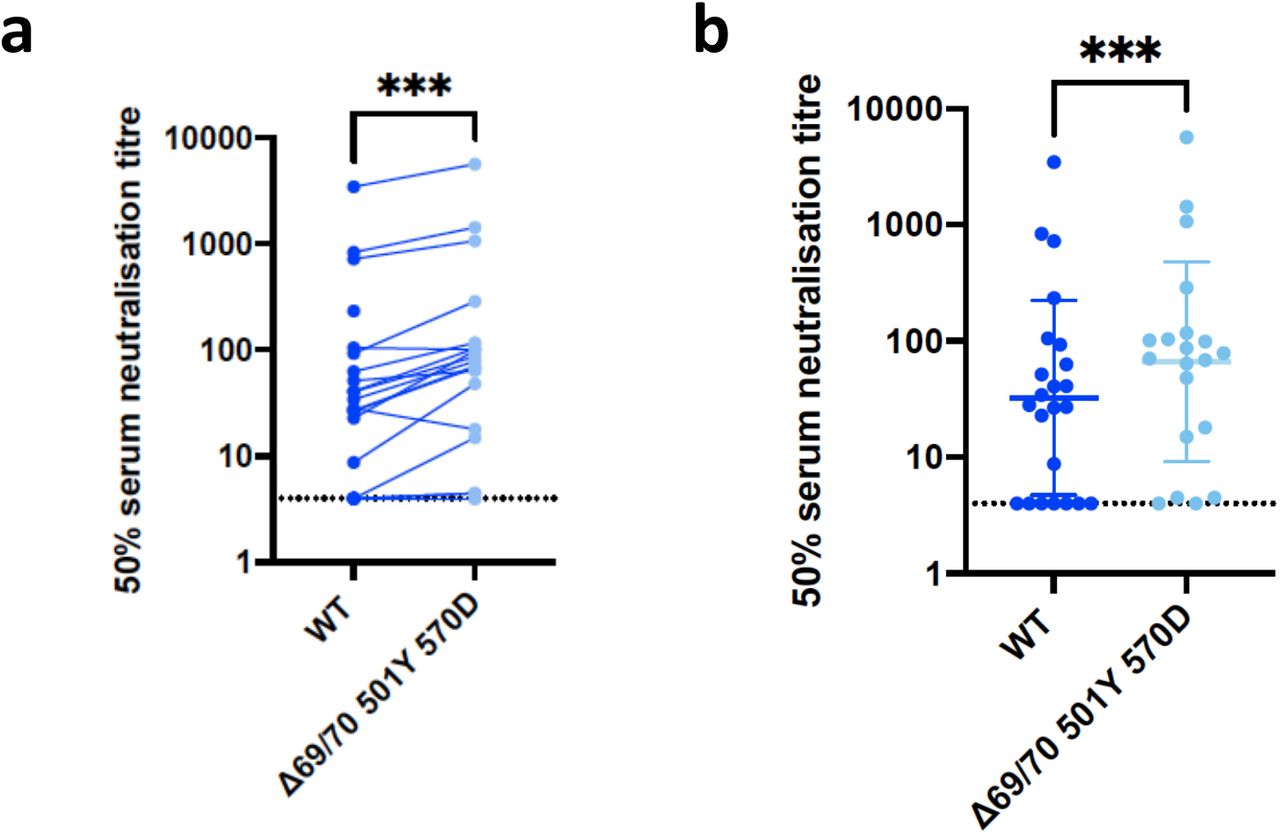
We next generated mutated pseudoviruses carrying S protein with mutations N501Y, A570D and the H69/V70 deletion. We observed no reduction in the ability of sera from vaccinees to inhibit either WT or mutant virus (Extended Data Fig. 2). In fact, we observed that vaccine sera displayed higher neutralizing activity against the N501Y, A570D and H69/V70 deletion mutant relative to WT virus. Next, we then tested a panel of sera from 11 recovered individuals and found that these sera also neutralised both wild type and the mutated viruses similarly (Extended Data Fig. 3). The findings for vaccine sera may be related to a potential allosteric effect of ΔH69/V70 that might enhance neutralization by antibodies targeting cryptic sites on the S, such as the RBD site II (also named as class 4 site).
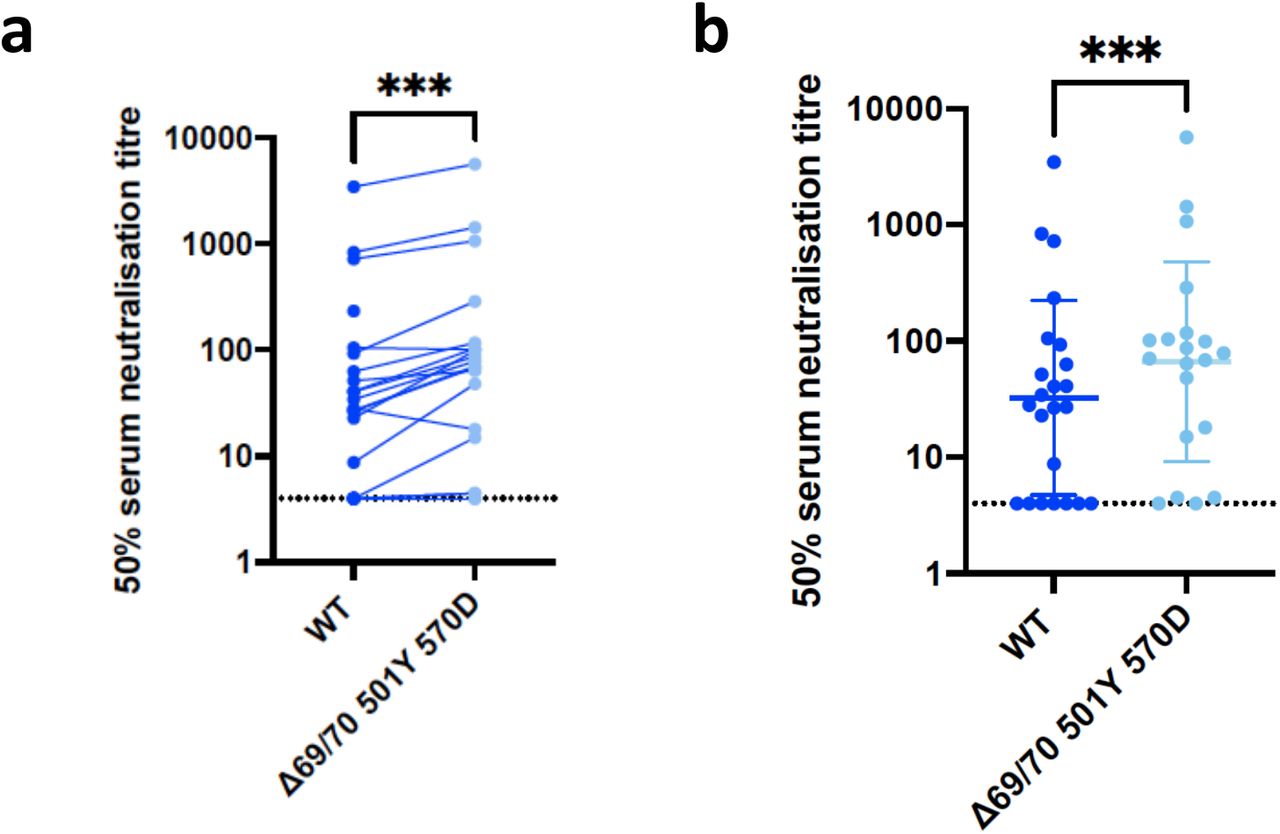
Neutralisation by vaccine sera against wild type and mutant SARS-CoV-2 pseudotyped viruses: (a-b) WT and Spike mutant with N501Y, A570D, ΔH69/V70. Data are representative of 2 independent experiments.

Indicated is serum log10 inverse dilution against % neutralisation. Where a curve is shifted to the right this indicates the virus is less sensitive to the neutralising antibodies in the serum. Data points represent means of technical replicates and error bars represent standard error of the mean.
We then generated the full set of eight mutations in the S protein present in B.1.1.7 variant (Fig. 1b and Table 1), ΔH69/V70, Δ144, N501Y and A570D in the S1 subunit and P681H, T716I, S982A and D1118H in the S2 subunit. All constructs also contained D614G. We found that among 15 individuals with neutralisation activity three weeks after receiving a single dose of the the BNT162b2 mRNA vaccine, 10 showed evidence of reduction in efficacy of antibodies against the B.1.1.7 mutant (Fig. 1c-d). The highest fold change was approximately 6. Amongst sera from 7 recovered individuals, only 2 demonstrated reduced potency against B.1.1.7 (Fig. 1e-h and Extended Data Fig. 4).

Indicated is serum log10 inverse dilution against % neutralisation. Where a curve is shifted to the right this indicates the virus is less sensitive to the neutralising antibodies in the serum. Data points represent means of technical replicates and error bars represent standard error of the mean.
B.1.1.7 variant escapes from NTD- and RBM-specific mAb-mediated neutralization
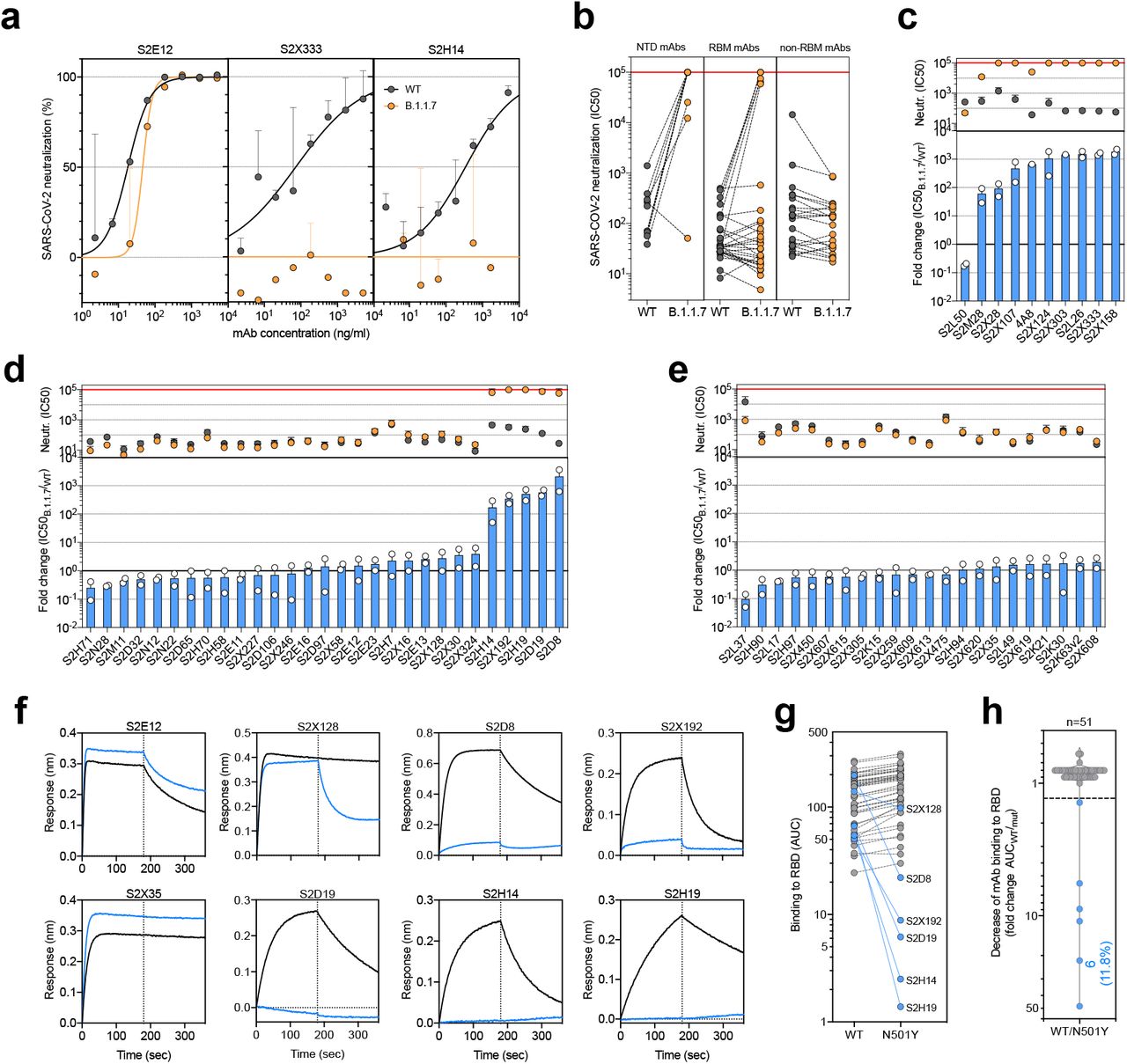
To investigate the role of the full set of mutations in NTD, RBD and S2 present in the B.1.1.7 variant, we tested 61 mAbs isolated from 15 individuals that recovered from WT SARS-CoV-2 infection with an in-vitro pseudotyped neutralization assay using VeroE6 target cells expressing TMPRSS2 (Extended Data Table 1). We found that 20 out of 61 (32.8%) mAbs showed a greater than 2-fold loss of neutralizing activity of B.1.1.7 variant compared to WT
SARS-CoV-2 (Fig. 2a,b and Extended Data Fig. 5). Remarkably, the B.1.1.7 mutant virus was found to fully escape neutralization by 7 out of 10 NTD-targeting mAbs (70%), and partial escape from additional 2 mAbs (20%) (Fig. 2c). We previously showed that the deletion of residue 144 abrogates binding by 4 out of 6 NTD-specific mAbs tested, possibly accounting for viral neutralization escape by most NTD-specific antibodies22. Of the 29 RBM-targeting mAbs, 5 (17.2%) showed more then 100-fold decrease in B.1.1.7 neutralization, and additional 6 mAbs (20.7%) had a partial 2-to-10-fold reduction (Fig. 2d). Finally, all RBD-specific non-RBM-targeting mAbs tested fully retained B.1.1.7 neutralizing activity (Fig. 2e).

a-c, Neutralisation of WT (black) and B.1.1.7 (orange) SARS-CoV-2-MLV by 9 NTD-targeting (a), 27 RBM-targeting (b) and 22 non-RBM-targeting (c) mAbs.
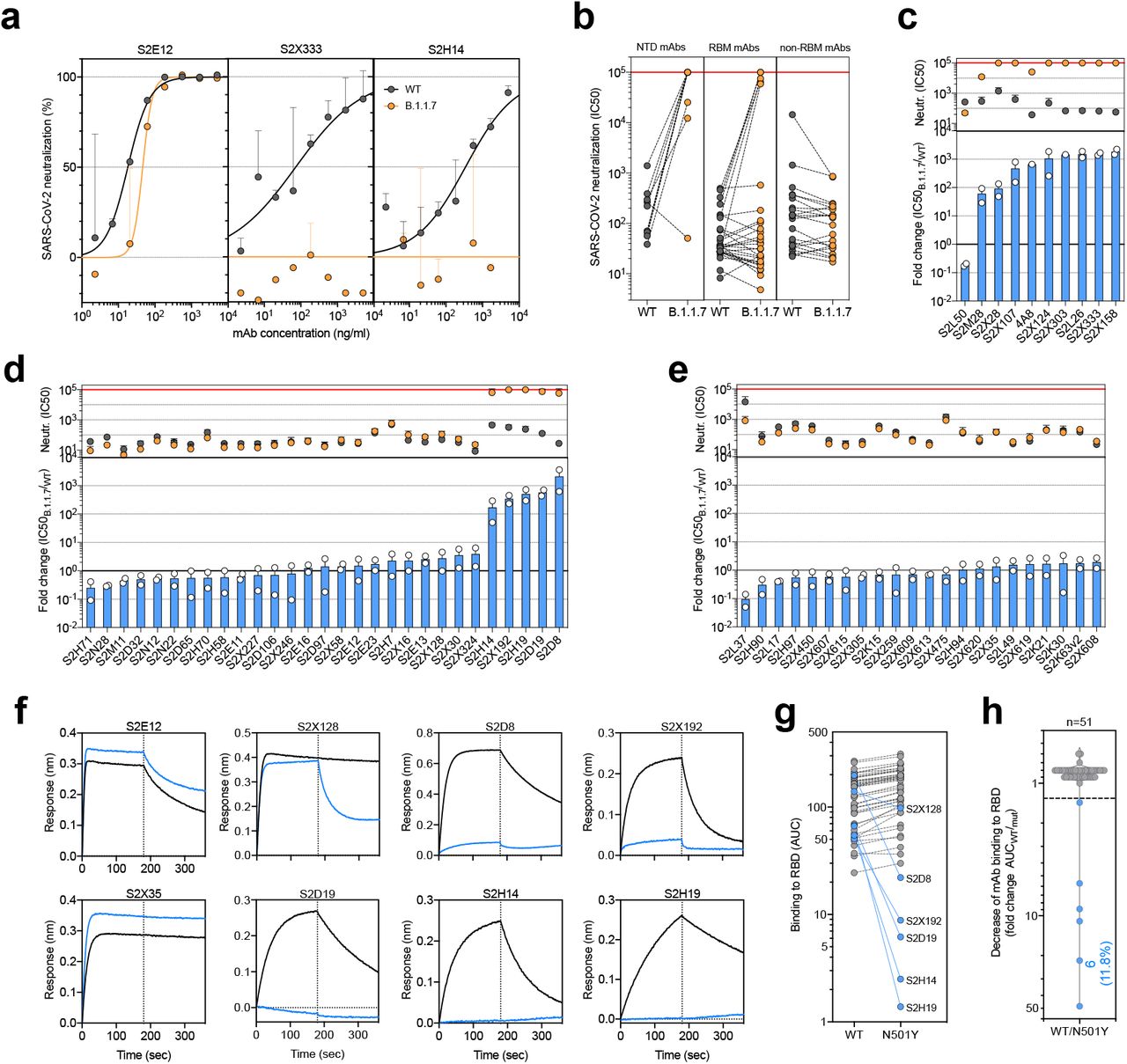
a, Neutralisation of WT (black) and B.1.1.7 (orange) Spike pseudotyped SARS-CoV-2-MLV by 3 selected mAbs (S2E12, S2×333 and S2H14) from one representative experiment. Shown is the mean ± s.d. of 2 technical replicates. b, Neutralisation of WT and B.1.1.7 SARS-CoV-2-MLV by 61 mAbs targeting NTD (n=10), RBM (n=29) and non-RBM sites in the RBD (n=22). Shown are the mean IC50 values (ng/ml) of n=2 independent experiments. c-e, Neutralisation shown as mean IC50 values (upper panel) and mean fold change of B.1.1.7 relative to WT (lower panel) of NTD (c), RBM (d) and non-RBM (e) mAbs. Lower panel shows IC50 values from 2 independent experiments. f-h, Kinetics of binding of mAbs to WT (black) and N501Y (blue) RBD as measured by bio-layer interferometry (BLI). Shown in (f) are the 6 RBM-targeting mAbs with reduced binding to N501Y RBD and 2 representative mAbs (RBM-targeting S2E12 and non-RBM-targeting S2×35). Area under the curve (AUC) (g) and AUC fold change (h) of 51 mAbs tested against WT and N501Y RBD. mAbs with a >1.3 AUC fold change shown in blue.
To address the role of B.1.1.7 N501Y mutation in the neutralization escape from RBM-specific antibodies, we tested the binding of 51 RBD-specific mAbs to WT and N501Y mutant RBD by biolayer interferometry (Fig. 2f and Extended Data Fig. 6). The 5 RBM-specific mAbs that failed to neutralize B.1.1.7 variant (Fig. 2d) showed a complete loss of binding to N501Y RBD mutant (Fig. 2g, h), demonstrating a critical role for this mutation as an escape mechanism for RBM-targeting mAbs.

a-b, Binding to WT (black) and N501Y (blue) RBD by 22 RBM-targeting (a) and 21 non-RBM-targeting (b) mAbs. An antibody of irrelevant specificity was included as negative control.
The decreased neutralizing activity of the immune sera from vaccinees and convalescent patients against B.1.1.7, but not against Δ69/70-501Y-570D mutant (Fig. 1 and Extended Data Fig. 2), could be the result of a loss of neutralizing activity of both RBD- and NTD-targeting antibodies, and suggests that the key mutations driving polyclonal escape are Δ144 and N501Y.
SARS-CoV-2 B.1.1.7 binds human ACE2 with higher affinity than WT
SARS-CoV-2 and SARS-CoV enter host cells through binding of the S glycoprotein to ACE21,23. Previous studies showed that the binding affinity of SARS-CoV for human ACE2 correlated with the rate of viral replication in distinct species, transmissibility and disease severity 24–26. To understand the potential contribution of receptor interaction to infectivity, we set out to evaluate the influence of the B.1.1.7 RBD substitution N501Y on receptor engagement. We used biolayer interferometry to study binding kinetics and affinity of the purified human ACE2 ectodomain (residues 1-615) to immobilized biotinylated SARS-CoV-2 B.1.1.7 or WT RBDs. We found that ACE2 bound to the B.1.1.7 RBD with an affinity of 22 nM compared to 133 nM for the WT RBD (Extended Data Fig. 7), in agreement with our previous deep-mutational scanning measurements using dimeric ACE227. Although ACE2 bound with comparable on-rates to both RBDs, the observed dissociation rate constant was slower for B.1.1.7 than for the WT RBD (Table 2). Enhanced binding of the B.1.1.7 RBD to human ACE2 resulting from the N501Y mutation might participate to the efficient ongoing transmission of this newly emerged SARS-CoV-2 lineage, and possibly reduced opportunity for antibody binding.

a-b. BLI binding analysis of the human ACE2 ectodomain (residues 1-615) to immobilized SARS-CoV-2 WT RBD (a) and B.1.1.7 RBD (b). Black lines correspond to a global fit of the data using a 1:1 binding model.
Kinetic analysis of human ACE2 binding to SARS-CoV-2 B.1.1.7 and Wuhan-1 RBDs by biolayer interferometry. Values reported represent the global fit to the data shown in Extended Data Fig. 7.
Further loss of neutralization of vaccine-elicited antibodies against a B.1.1.7 Spike carrying E484K mutation
The E484K substitution (Fig 3a) has been identified as antigenically important, being reported as an escape mutation for several monoclonal antibodies including C121, C144, REGN10933 and REGN10934. E484K is also known to be present in the B.1.351 (501Y.V2) and the P.1 (501Y.V3) lineages in combination with amino acid replacements at N501 and K417. As of 1st Feb 2021, fourteen English and two Welsh B.1.1.7 sequences from viral isolates contained the E484K substitution (Fig. 3b), The number of B.1.1.7 sequences has been increasing since the start of December 2020 (Fig. 3c). Phylogenetic analysis suggests that there were two independent acquisitions in English and Welsh sequences (Fig. 3b).

a. Representation of Spike RBM:ACE2 interface (PDB: 6M0J) with residues E484, N501 and K417 highlighted as spheres coloured by element b. Maximum likelihood phylogeny of a subset of sequences from the United Kingdom bearing the E484K mutation (green) and lineage B.1.1.7 (blue). As of 1st Feb 2021, 16 sequences from the B.1.1.7 lineage (two unrelated sequences in Wales, and one cluster of 14 in England) have acquired the E484K mutation (red). c. Sequence accumulation over time in GISAID for sequences with B.1.1.7 and E484K.
We generated pseudoviruses bearing B.1.1.7 Spike mutations with or without additional E484K and tested these against vaccine sera. We observed a significant loss of neutralising activity compared with WT and with B.1.1.7; mean fold change for the E484K B.1.1.7 Spike was 9.6 compared to 2.4 for B.1.1.7 alone (p<0.05) (Fig. 4).

Neutralisation potency of vaccine sera against pseudovirus virus bearing Spike mutations in the B1.1.7 lineage with and without E484K in the receptor binding domain (all In Spike D614G background). a, Neutralisation curves for five vaccinated individuals. Data points represent mean of technical replicates with standard error and are representative of two independent experiments b, c. Indicated is 50% neutralisation titre for each virus expressed as fold change relative to WT. WT (n=24), B.1.1.7 (n=24), B.1.1.7 +E484K (n=12). In c only data for participants with FC from three viruses are presented. Data points represent means of two independent experiments.
Discussion
Serum neutralizing activity is a correlate of protection for other respiratory viruses, including influenza28 and respiratory syncytial virus where prohylaxis with monoclonal antibodies has been used in at-risk groups29,30. Neutralising antibody titres appeared to be highly correlated with vaccine protection against SARS-CoV-2 rechallenge in non-human primates, and importantly, there was no correlation between T cell responses (as measured by ELISPOT) and protection31. Moreover, passive transfer of purified polyclonal IgGs from convalescent macaques protected naïve macaques against subsequent SARS-CoV-2 challenge32. Coupled with multiple reports of re-infection, there has therefore been significant attention placed on virus neutralisation.
This study reports on the neutralisation escape from sera collected after one dose of the BNT162b2 vaccine. The participants of this study had a median age of over 80, in line with the targeting of this age group in the initial rollout of the vaccination campaign in the UK. Participants showed similar neutralising activity against wild type pseudovirus as in the phase I/II study, with geometric mean titres of 24 and 29, respectively12. The three mutations in S (N501Y, A570D, ΔH69/V70) did not appear to impact neutralisation in a pseudovirus assay. However, we demonstrated that a pseudovirus bearing S protein with the full set of mutations present in the B.1.1.7 variant (i.e., ΔH69/V70, Δ144, N501Y, A570D, P681H, T716I, S982A, D1118H) did result in small reduction in neutralisation by sera from vaccinees. A reduction in neutralization titres from mRNA-elicited antibodies in volunteers who received two doses (using both mRNA-1273 and BNT162b2 vaccines) was also observed by Wang et al.33 using pseudoviruses carrying the N501Y mutation. Another study reported on a modest and not significant (average 1.2 fold) reduction of neutralization against the B.1.1.7 variant in sera from individuals vaccinated with two doses of mRNA-127334. The level of neutralizing antibodies observed in both of these studies was approximately one log higher than the one observed in the cohort of vaccinees described in this study. This may be related to the older age of the individuals from this study as well as to the fact that these were exposed to only one dose of the BNT162b2 vaccine. In general, it is expected that the effect of mutations on the neutralization by polyclonal serum antibodies might be more prominent on low-titre in contrast to high-titre sera. It is important to study virus neutralisation at lower serum neutralisation titres because decline in neutralisation titres over time is expected to occur following vaccination.
The reduced neutralizing activity observed with polyclonal antibodies elicited by mRNA vaccines observed in this study and in Wang et al.33 is further supported by the loss of neutralizing activity observed with human mAbs directed to both RBD and, to a major extent, to NTD. In the study by Wang et al., 6 out 17 RDB-specific mAbs isolated from mRNA-1273 vaccinated individuals showed more than 100-fold neutralization loss against N501Y mutant, a finding that is consistent with the loss of neutralization by 5 out 29 RBM-specific mAbs described in this study.
Multiple variants, including the 501Y.V2 and B.1.1.7 lineages, harbor multiple mutations as well as deletions in NTD, most of which are located in a site of vulnerability that is targeted by all known NTD-specific neutralizing antibodies22. The role of NTD-specific neutralizing antibodies might be under-estimated, in part by the use of neutralization assays based on target cells over-expressing ACE2 receptor. NTD-specific mAbs were suggested to interfere with viral entry based on other accessory receptors, such as DC-SIGN and L-SIGN35, and their neutralization potency was found to be dependent on different in vitro culture conditions22. The observation that 9 out of 10 NTD-specific neutralizing antibodies failed to show a complete or near-complete loss of sneutralizing activity against B.1.1.7 indicates that this new variant may have evolved also to escape from this class of antibodies, that may have a yet unrecognized role in protective immunity. Wibmer et al.36 have also recently reported the loss of neutralization of 501Y.V2 by the NTD-specific mAb 4A8, likely driven by the R246I mutation. This result is in line with the lack of neutralization of B.1.1.7 by 4A8 mAb observed in this study, likely caused by Δ144 due to loss of binding22. Finally, the role of NTD mutations (in particular, L18F, Δ242-244 and R246I) was further supported by the marked loss of neutralization observed by Wibmer et al.36 against 501Y.V2 compared to the chimeric virus carrying only the RBD mutations K417N, E484K and N501Y. Taken together, the presence of multiple escape mutations in NTD is supportive of the hypothesis that this region of the Spike, in addition to RBM, is also under immune pressure.
E484K has been shown to impact neutralisation by monoclonal antibodies or convalescent sera, especially in combination with N501Y and K417N16,37–39. Worryingly, we have shown that there are multiple B.1.1.7 sequences in the UK bearing E484K with early evidence of transmission as well as independent aquisitions. We measured further reduction neutralisation titers by vaccine sera when E484K was present alongside the B.1.1.7 S mutations. Consistent with our findings, Wu and co-authors34 have shown that variants carrying the E484K mutation resulted in 3-to-6 fold reduction in neutralization by sera from mRNA-1273 vaccinated individuals.
Evidence for the importance role of NTD deletions in combination with E484K in immune escape is provided by Andreano et al. who describe the emergence of Δ140 in virus co-incubated with potently neutralizing convalescent plasma, causing a 4-fold reduction in neutralization titre. This Δ140 mutant subsequently acquired E484K which resulted in a further 4-fold drop in neutralization titre indicating a two residue change across NTD and RBD represents an effective pathway of escape that can dramatically inhibit the polyclonal response.
Our study was limited by relatively small sample size. Although the Spike pseudotyping system has been shown to faithfully represent full length infectious virus, there may be determinants outside the S that influence escape from antibody neutralization either directly or indirectly in a live replication competent system. On the other hand live virus systems allow replication and therefore mutation to occur, and rigorous sequencing at multiple steps is needed.
Amidst high transmission in many parts of the world, vaccines are a key part of a long term strategy to bring SARS-CoV-2 under control. Our data suggest that vaccine escape will be inevitable in the future, and should be mitigated by designing next generation vaccines with mutated S sequences and using alternative viral antigens. Currently however, vaccines are likely to contribute controlling SARS-CoV-2 infections in the short term.
Data Availability
Data are available from the corresponding author on request
Author contributions
Conceived study: D.C., RKG, DAC. Designed study and experiments: RKG, DAC, LEM, JB, MW, JT, LCG, GBM, RD, BG, NK, AE, M.P., D.V., L.P., A.D.M, J.B., D.C. Performed experiments: BM, DAC, RD, IATMF, LCG, GBM. Interpreted data: RKG, DAC, BM, RD, IATMF, LEM, JB, KGCS. A.D.M., J.B. and C.S.F. carried out pseudovirus neutralization assays. D.P. produced pseudoviruses. M.S.P., L.P., D.V. and D.C. designed the experiments. A.C.W., N.S. and S.J. expressed and purified the proteins. K.C., S.J. and E.C. sequenced and expressed antibodies. E.C. and K.C. performed mutagenesis for mutant expression plasmids. A.C.W. M.A.T., J.E.B., and S.B. performed binding assays. A.R., A.F.P and C.G contributed to donor’s recruitment and sample collection related to mAbs isolation. H.W.V., G.S., A.L., D.V., L.P. and D.C. analyzed the data and prepared the manuscript with input from all authors.
Competing interests
A.D.M., J.B., D.P., C.S.F., S.B., K.C., N.S., E.C., G.S., S.J., A.L., H.W.V., M.S.P., L.P. and D.C. are employees of Vir Biotechnology and may hold shares in Vir Biotechnology. H.W.V. is a founder of PierianDx and Casma Therapeutics. Neither company provided funding for this work or is performing related work. D.V. is a consultant for Vir Biotechnology Inc. The Veesler laboratory has received a sponsored research agreement from Vir Biotechnology Inc. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. RKG has received consulting fees from UMOVIS Lab, Gilead and ViiV.
MATERIALS AND METHODS
Participant recruitment and ethics
Participants who had received the first dose of vaccine and individuals with COVID-19 were consented into the Covid-19 cohort of the NIHR Bioresource. The study was approved by the East of England – Cambridge Central Research Ethics Committee (17/EE/0025).
SARS-CoV-2 serology by multiplex particle-based flow cytometry (Luminex)
Recombinant SARS-CoV-2 N, S and RBD were covalently coupled to distinct carboxylated bead sets (Luminex; Netherlands) to form a 3-plex and analyzed as previously described (Xiong et al. 2020). Specific binding was reported as mean fluorescence intensities (MFI).
Recombinant expression of SARS-CoV-2-specific mAbs
Human mAbs were isolated from plasma cells or memory B cells of SARS-CoV-2 immune donors, as previously described 40–42. Recombinant antibodies were expressed in ExpiCHO cells at 37°C and 8% CO2. Cells were transfected using ExpiFectamine. Transfected cells were supplemented 1 day after transfection with ExpiCHO Feed and ExpiFectamine CHO Enhancer. Cell culture supernatant was collected eight days after transfection and filtered through a 0.2 µm filter. Recombinant antibodies were affinity purified on an ÄKTA xpress FPLC device using 5 mL HiTrap™MabSelect™PrismA columns followed by buffer exchange to Histidine buffer (20 mM Histidine, 8% sucrose, pH 6) using HiPrep 26/10 desalting columns
Generation of S mutants
Amino acid substitutions were introduced into the D614G pCDNA_SARS-CoV-2_S plasmid as previously described43 using the QuikChange Lightening Site-Directed Mutagenesis kit, following the manufacturer’s instructions (Agilent Technologies, Inc., Santa Clara, CA). Sequences were checked by Sanger sequencing.
Preparation of B.1.1.7 SARS-CoV-2 S glycoprotein-encoding-plasmid used to produce SARS-CoV-2-MLV based on overlap extension PCR. Briefly, a modification of the overlap extension PCR protocol44 was used to introduce the nine mutations of the B.1.1.7 lineage in the SARS-CoV-2 S gene. In a first step, 9 DNA fragments with overlap sequences were amplified by PCR from a plasmid (phCMV1, Genlantis) encoding the full-length SARS-CoV-2 S gene (BetaCoV/Wuhan-Hu-1/2019, accession number mn908947). The mutations (del-69/70, del-144, N501Y, A570D, D614G, P681H, S982A, T716I and D1118H) were introduced by amplification with primers with similar Tm. Deletion of the C-terminal 21 amino acids was introduced to increase surface expression of the recombinant S45. Next, 3 contiguous overlapping fragments were fused by a first overlap PCR (step 2) using the utmost external primers of each set, resulting in 3 larger fragments with overlapping sequences. A final overlap PCR (step 3) was performed on the 3 large fragments using the utmost external primers to amplify the full-length S gene and the flanking sequences including the restriction sites KpnI and NotI. This fragment was digested and cloned into the expression plasmid phCMV1. For all PCR reactions the Q5 Hot Start High fidelity DNA polymerase was used (New England Biolabs Inc.), according to the manufacturer’s instructions and adapting the elongation time to the size of the amplicon. After each PCR step the amplified regions were separated on agarose gel and purified using Illustra GFX™PCR DNA and Gel Band Purification Kit (Merck KGaA).
Pseudotype virus preparation
Viral vectors were prepared by transfection of 293T cells by using Fugene HD transfection reagent (Promega). 293T cells were transfected with a mixture of 11ul of Fugene HD, 1µg of pCDNAΔ19Spike-HA, 1ug of p8.91 HIV-1 gag-pol expression vector46,47, and 1.5µg of pCSFLW (expressing the firefly luciferase reporter gene with the HIV-1 packaging signal). Viral supernatant was collected at 48 and 72h after transfection, filtered through 0.45um filter and stored at −80°C. The 50% tissue culture infectious dose (TCID50) of SARS-CoV-2 pseudovirus was determined using Steady-Glo Luciferase assay system (Promega).
Serum/plasma pseudotype neutralization assay
Spike pseudotype assays have been shown to have similar characteristics as neutralisation testing using fully infectious wild type SARS-CoV-220. Virus neutralisation assays were performed on 293T cell transiently transfected with ACE2 and TMPRSS2 using SARS-CoV-2 Spike pseudotyped virus expressing luciferase48. Pseudotyped virus was incubated with serial dilution of heat inactivated human serum samples or sera from vaccinees in duplicate for 1h at 37°C. Virus and cell only controls were also included. Then, freshly trypsinized 293T ACE2/TMPRSS2 expressing cells were added to each well. Following 48h incubation in a 5% CO2 environment at 37°C, luminescence was measured using the Steady-Glo Luciferase assay system (Promega).
IFNγ FLUOROSPOT assays
Frozen PBMCs were rapidly thawed, and the freezing medium was diluted into 10ml of TexMACS media (Miltenyi Biotech), centrifuged and resuspended in 10ml of fresh media with 10U/ml DNase (Benzonase, Merck-Millipore via Sigma-Aldrich), PBMCs were incubated at 37°C for 1h, followed by centrifugation and resuspension in fresh media supplemented with 5% Human AB serum (Sigma Aldrich) before being counted. PBMCs were stained with 2ul of each antibody: anti-CD3-fluorescein isothiocyanate (FITC), clone UCHT1; anti-CD4-phycoerythrin (PE), clone RPA-T4; anti-CD8a-peridinin-chlorophyll protein - cyanine 5.5 (PerCP Cy5.5), clone RPA-8a (all BioLegend, London, UK), LIVE/DEAD Fixable Far Red Dead Cell Stain Kit (Thermo Fisher Scientific). PBMC phenotyping was performed on the BD Accuri C6 flow cytometer. Data were analysed with FlowJo v10 (Becton Dickinson, Wokingham, UK). 1.5 to 2.5 x 105 PBMCs were incubated in pre-coated Fluorospot plates (Human IFNγ FLUOROSPOT (Mabtech AB, Nacka Strand, Sweden)) in triplicate with peptide mixes specific for Spike, Nucleocapsid and Membrane proteins of SARS-CoV-2 (final peptide concentration 1µg/ml/peptide, Miltenyi Biotech) and an unstimulated and positive control mix (containing anti-CD3 (Mabtech AB), Staphylococcus Enterotoxin B (SEB), Phytohaemagglutinin (PHA) (all Sigma Aldrich)) at 37°C in a humidified CO2 atmosphere for 48 hours. The cells and medium were decanted from the plate and the assay developed following the manufacturer’s instructions. Developed plates were read using an AID iSpot reader (Oxford Biosystems, Oxford, UK) and counted using AID EliSpot v7 software (Autoimmun Diagnostika GmbH, Strasberg, Germany). All data were then corrected for background cytokine production and expressed as SFU/Million PBMC or CD3 T cells.
MAbs pseudovirus neutralization assay
MLV-based SARS-CoV-2 S-glycoprotein-pseudotyped viruses were prepared as previously described (Pinto et al., 2020). HEK293T/17cells were cotransfected with a WT or B.1.1.7 SARS-CoV-2 Spike glycoprotein-encoding-plasmid, an MLV Gag-Pol packaging construct and the MLV transfer vector encoding a luciferase reporter using X-tremeGENE HP transfection reagent (Roche) according to the manufacturer’s instructions. Cells were cultured for 72 h at 37°C with 5% CO2 before harvesting the supernatant. VeroE6 stably expressing human TMPRSS2 were cultured in DMEM containing 10% FBS, 1% penicillin– streptomycin, 8 µg/mL puromycin and plated into 96-well plates for 16–24 h. Pseudovirus with serial dilution of mAbs was incubated for 1 h at 37°C and then added to the wells after washing 2 times with DMEM. After 2–3 h DMEM containing 20% FBS and 2% penicillin– streptomycin was added to the cells. Following 48-72 h of infection, Bio-Glo (Promega) was added to the cells and incubated in the dark for 15 min before reading luminescence with Synergy H1 microplate reader (BioTek). Measurements were done in duplicate and relative luciferase units were converted to percent neutralization and plotted with a non-linear regression model to determine IC50 values using GraphPad PRISM software (version 9.0.0).
Antibody binding measurements using bio-layer interferometry (BLI)
MAbs were diluted to 3 µg/ml in kinetic buffer (PBS supplemented with 0.01% BSA) and immobilized on Protein A Biosensors (FortéBio). Antibody-coated biosensors were incubated for 3□min with a solution containing 5□µg□/ml of WT or N50Y SARS-CoV-2 RBD in kinetic buffer, followed by a 3-min dissociation step. Change in molecules bound to the biosensors caused a shift in the interference pattern that was recorded in real time using an Octet RED96 system (FortéBio). The binding response over time was used to calculate the area under the curve (AUC) using GraphPad PRISM software (version 9.0.0).
Production of SARS-CoV-2 and B.1.1.7 receptor binding domains and human ACE2
The SARS-CoV-2 RBD (BEI NR-52422) construct was synthesized by GenScript into CMVR with an N-terminal mu-phosphatase signal peptide and a C-terminal octa-histidine tag (GHHHHHHHH) and an avi tag. The boundaries of the construct are N-328RFPN331 and 528KKST531-C49. The B.1.1.7 RBD was synthesized by GenScript into pCMVR with the same boundaries and construct details with a mutation at N501Y. These plasmids were transiently transfected into Expi293F cells using Expi293F expression medium (Life Technologies) at 37°C 8% CO2 rotating at 150 rpm. The cultures were transfected using PEI cultivated for 5 days. Supernatants were clarified by centrifugation (10 min at 4000xg) prior to loading onto a nickel-NTA column (GE). Purified protein was biotinylated overnight using BirA (Avidity) prior to SEC into PBS. Human ACE2-Fc (residues 1-615 with a C-terminal thrombin cleavage site and human Fc tag) were synthesized by Twist. Clarified supernatants were affinity purified using a Protein A column (GE LifeSciences) directly neutralized and buffer exchanged. The Fc tag was removed by thrombin cleavage in a reaction mixture containing 3 mg of recombinant ACE2-FC ectodomain and 10 μg of thrombin in 20 mM Tris-HCl pH8.0, 150 mM NaCl and 2.5 mM CaCl2.The reaction mixture was incubated at 25°C overnight and re-loaded on a Protein A column to remove uncleaved protein and the Fc tag. The cleaved protein was further purified by gel filtration using a Superdex 200 column 10/300 GL (GE Life Sciences) equilibrated in PBS.
Protein affinity measurement using bio-layer interferometry
Biotinylated RBD (either WT or N501Y) were immobilized at 5 ng/uL in undiluted 10X Kinetics Buffer (Pall) to SA sensors until a load level of 1.1nm, A dilution series of either monomeric ACE2 or Fab in undiluted kinetics buffer starting at 1000-50nM was used for 300-600 seconds to determine protein-protein affinity. The data were baseline subtracted and the plots fitted using the Pall FortéBio/Sartorius analysis software (version 12.0). Data were plotted in Prism.
Acknowledgements
We would like to thank Cambridge University Hospitals NHS Trust Occupational Health Department. We would also like to thank the NIHR Cambridge Clinical Research Facility and staff at CUH and. We would like to thank Eleanor Lim and Georgina Okecha. We thank Dr James Voss for the kind gift of HeLa cells stably expressing ACE2. RKG is supported by a Wellcome Trust Senior Fellowship in Clinical Science (WT108082AIA). LEM is supported by a Medical Research Council Career Development Award (MR/R008698/1). SAK is supported by the Bill and Melinda Gates Foundation via PANGEA grant: OPP1175094. DAC is supported by a Wellcome Trust Clinical PhD Research Fellowship. KGCS is the recipient of a Wellcome Investigator Award (200871/Z/16/Z). This research was supported by the National Institute for Health Research (NIHR) Cambridge Biomedical Research Centre, the Cambridge Clinical Trials Unit (CCTU), and the NIHR BioResource. The views expressed are those of the authors and not necessarily those of the NIHR or the Department of Health and Social Care. JAGB is supported by the Medical Research Council (MC_UP_1201/16). IATM is funded by a SANTHE award.
Footnotes
Substantial revision of the previous manuscript with more serological data as well as analysis of the effect of B.1.1.7 on a large panel of human monoclonal neutralizing antibodies




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}