
Abstract
Objective The aim of this study was to search for genes/variants that modify the effect of LRRK2 mutations in terms of penetrance and age-at-onset of Parkinson’s disease.
Methods We performed the first genome-wide association study of penetrance and age-at-onset of Parkinson’s disease in LRRK2 mutation carriers (776 cases and 1,103 non-cases at their last evaluation). Cox proportional hazard models and linear mixed models were used to identify modifiers of penetrance and age-at-onset of LRRK2 mutations, respectively. We also investigated whether a polygenic risk score derived from a published genome-wide association study of Parkinson’s disease was able to explain variability in penetrance and age-at-onset in LRRK2 mutation carriers.
Results A variant located in the intronic region of CORO1C on chromosome 12 (rs77395454; P-value=2.5E-08, beta=1.27, SE=0.23, risk allele: C) met genome-wide significance for the penetrance model. A region on chromosome 3, within a previously reported linkage peak for Parkinson’s disease susceptibility, showed suggestive associations in both models (penetrance top variant: P-value=1.1E-07; age-at-onset top variant: P-value=9.3E-07). A polygenic risk score derived from publicly available Parkinson’s disease summary statistics was a significant predictor of penetrance, but not of age-at-onset.
Interpretation This study suggests that variants within or near CORO1C may modify the penetrance of LRRK2 mutations. In addition, common Parkinson’s disease associated variants collectively increase the penetrance of LRRK2 mutations.
Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disease in older adults.1 Several genes showing autosomal dominant (SNCA, LRRK2, VPS35) or recessive (PRKN, PINK1, DJ-1) inheritance patterns have been identified as the cause of familial PD. These genes harbor rare, high penetrance mutations that explain up to 10% of familial PD cases in different populations.1,2 Recently, large genome-wide association studies (GWAS) have identified over 90 loci with small individual effects on disease risk in both familial and sporadic PD.3,4
Mutations in LRRK2 are among the most common genetic causes of PD.1,2 The most frequent mutation is G2019S (rs34637584), which explains up to 10% of familial PD cases and 1-2% of all PD cases.2,5 Among PD patients, the frequency of the G2019S mutation is approximately 3% in Europeans, 16-19% in Ashkenazi Jews and up to 42% in Arab-Berbers.6-14 Estimates of the risk for developing PD among LRRK2 G2019S mutation carriers range from 15% to 85%.15-18 To explain the incomplete penetrance of G2019S, it has long been hypothesized that there are other genes/variants outside of LRRK2 acting to modify its effect (LRRK2 modifiers). Identification of LRRK2 modifiers could aid the development of novel prevention and treatment strategies for PD.
Most studies of LRRK2 modifiers, to date, have focused on candidate genes. Since the protein product of LRRK2 may interact with α-synuclein (encoded by SNCA), and tau (encoded by MAPT),19,20 variants in SNCA and MAPT were widely investigated. However, the results have been inconsistent, possibly due to small sample sizes and differences in variants and populations investigated.21-29 Other PD associated genes such as GBA,28 BST1,28 GAK,29 and PARK1628,30,31 have also been investigated. However, the number of studies is limited and findings remain to be replicated. Genome-wide searches for LRRK2 modifiers are sparse and limited to linkage studies. Using 85 LRRK2 carriers from 38 families, a genome-wide linkage study of LRRK2 modifiers found a suggestive linkage region at 1q32 (LOD=2.43); but that study did not identify any candidate genes/variants underlying the linkage peak.32 A genome-wide linkage scan in Arab-Berber PD families found DNM3 as a LRRK2 modifier.33 This finding was not independently replicated, although it was still significant in a meta-analysis including the participants reported in the original finding.24,34 Genome-wide association studies have successfully detected many disease genes/variants, including those associated with PD. However, to date, no GWAS for LRRK2 modifiers has been reported, probably due to limitations in sample size and corresponding statistical power.
In this study, we recruited LRRK2 mutation carriers from multiple centers and performed the first GWAS to identify genes/variants that modify the penetrance and age-at-onset of PD among LRRK2 mutation carriers. Using the largest cohort to date, which consisted of 1,879 LRRK2 mutation carriers (including 776 PD cases), one genome-wide significant association signal was found in the intronic region of the CORO1C gene. In addition, we found that a polygenic risk score (PRS) derived from publicly available PD GWAS summary statistics, was associated with penetrance, but not age-at-onset, of PD in LRRK2 mutation carriers.
Methods
Study participants
The studies and the LRRK2 mutation carriers were grouped into three cohorts. The first cohort was primarily identified from The Michael J. Fox Foundation’s LRRK2 Consortium and consisted of research sites worldwide (referred to as the MJFF consortium cohort). We searched PubMed and identified study groups that reported LRRK2 mutation carriers then asked them to participate in this study (PUBMED IDs: 16240353, 16333314, 18986508, and 16960813).35-38 We also made announcements at international conferences to recruit more study mutation carriers. Details can be found in their publications.35-38 To maximize participation and facilitate uniform data preparation across sites, a minimal dataset was submitted for all subjects that included LRRK2 mutation status, sex, age-at-onset (for PD cases), age at last evaluation (for non-PD participants) and pedigree information, along with the availability of a minimal amount of DNA (∼2 ug). The minimal phenotypic data were sent to Indiana University and the subjects were assigned a unique identifier. The second cohort was from Tel Aviv University, Israel (referred to as the Israel cohort). Participants were of Ashkenazi origin and recruited from the Movement Disorders Unit at Tel Aviv Medical Center. PD diagnosis was confirmed by a movement disorders specialist and clinical disease status (PD or not diagnosed as PD) was evaluated at the time of blood draw for genetic testing. The third cohort (referred to as the 23andMe cohort) consisted of research participants of the personal genetics company 23andMe, Inc. who were LRRK2 G2019S carriers and whose PD status was known. Individuals who reported via an online survey that they had been diagnosed with PD by a medical professional, were asked to provide their age at diagnosis. For individuals who affirmed at least once that they had not been diagnosed with PD, their age at the most recent completion of the survey was recorded. The study was approved by the Institutional review board at Indiana University; the Institutional Review Board (Helsinki) Committee of Tel Aviv Sourasky Medical Center and the National Helsinki Committee for Genetic Research in Humans, MINISTRY OF HEALTH, Israel; Ethical & Independent Review Services, a private institutional review board (http://www.eandireview.com).
Genotyping, quality review, and imputation
All study participants were genotyped on the Illumina Omni 2.5 Exome Array V1.1 (Illumina, San Diego, USA), except 166 participants from the Israel cohort, who were genotyped on an earlier version of the same array (V1.0). This array has common, rare, and exonic variants that were selected from diverse world population samples included in the 1000 Genomes Project. In total, there were >2.58M variants, including >567K exonic variants. Participants from the MJFF consortium and 23andMe were genotyped at the Center for Inherited Disease Research (CIDR) at Johns Hopkins University (Baltimore, MD, USA). The Israel cohort was genotyped at Tel Aviv University and two samples from the MJFF consortium were included for quality control. There were 134 duplicated and unexpected identical participants among all three cohorts. Pairwise concordance rates were all >99.97%, showing high consistency among the two genotyping labs and the two versions of the Illumina array.
Variants with genotypic missing rates >5% and non-polymorphic variants were excluded. In addition, variants with A/T or C/G alleles were also excluded due to strand ambiguity. Hardy-Weinberg equilibrium (HWE) was not used to filter variants because these participants were ascertained to be LRRK2 mutation carriers and this participant selection scheme would directly violate HWE and remove potential LRRK2 modifiers from the analysis.
To confirm the reported pedigree structure and detect cryptic relatedness, we used a set of 56,184 high quality (missing rate <2%, HWE P-values >0.001), common (MAF >0.1), and independent (linkage disequilibrium as measured by r2 <0.5) variants to calculate the pairwise identity by descent using PLINK.39 Reported pedigree structures were revised accordingly, if necessary. Mendelian error checking was performed in the revised pedigree structure. Any inconsistent genotypes were set to missing. The same set of variants was also used to estimate the principal components (PCs) of population stratification using Eigenstrat.40 All samples were imputed to the Haplotype Reference Consortium (http://www.haplotype-reference-consortium.org/) using Minimac3.41 A total of 725,802 high quality genotyped variants were selected for imputation (MAF >3%, HWE P-value >0.0001, and missing rate <5%). EAGLE v2.442 was used to phase genotyped variants for each sample. After filtering out variants with poor imputation quality score (R2 < 0.6) and checking for Mendelian inconsistencies using PLINK,39 a final dataset of 7,934,276 imputed and genotyped variants was used for association analyses.
Genome-wide association studies
Our association analysis tested two models: 1) variants modifying the penetrance for PD among LRRK2 mutation carriers (penetrance model), and 2) variants modifying the age-at-onset for PD among LRRK2 mutation carriers (age-at-onset model). For the penetrance model (including PD cases and those not diagnosed as PD at last evaluation), the association analysis was designed to identify variants associated with the time to PD diagnosis or last evaluation for undiagnosed mutation carriers. For the age-at-onset model (PD cases only), the association analysis tested whether variants contributed to the age-at-onset for PD cases among LRRK2 mutation carriers.
For the penetrance model, a mixed effect Cox proportional hazard model (frailty model) was used with sex, 10 PCs, array and cohort indicators as covariates. Family relationships were adjusted by using a kinship matrix calculated using R package COXME (https://cran.r-project.org/web/packages/coxme/index.html). For the age-at-onset model, a linear mixed model was fit with the same covariates as the penetrance model and a kinship matrix to adjust family relationships. Variants with MAF > 1% were tested for association in these two models. In addition to the single variant analyses, we performed gene-based association analyses for both the penetrance and age-at-onset models. We focused on rare exonic and splicing variants, based on annotations from Variant Effect Predictor (https://useast.ensembl.org/info/docs/tools/vep/index.html), and restricting to variants with MAF <3%. Only genotyped variants (N= 725,802) were used in the gene-based analyses due to the low quality of imputation for rare variants. The R package COXME was used to perform all analyses (https://cran.r-project.org/web/packages/coxme/index.html). Conditional analysis was conducted using the most significant variant in an associated region as a covariate, and additional signals within the associated region were determined based on P-values < 0.01.
Functional studies
To evaluate whether rs77395454 has immediate biological consequences on gene expression (eQTL) of nearby genes, we searched Open Targets Genetics (https://genetics.opentargets.org/) and GTEx (https://www.gtexportal.org/). In addition, protein-protein interaction (PPI) data was assessed to identify whether the protein product which maps to this locus either interacts directly with LRRK2 or has common interactors that are shared with LRRK2, using PINOT (Protein Interaction Network Online Tool) version 1.043 queried on 16th June 2020 (http://www.reading.ac.uk/bioinf/PINOT/PINOT_form.html).
Polygenic risk score analyses
In the largest GWAS analysis of PD susceptibility to date, Nalls et al meta-analyzed 17 datasets with 56,306 PD cases or proxy-cases and 1.4 million controls.4 Based on their results, they developed a PRS using summary statistics of 1,805 variants that can explain 26% of PD heritability.4 In this study, we performed PRS analysis using these 1,805 variants. Detailed information about how to select these 1,805 variants was described in Nalls et al.4 Since we were searching for LRRK2 modifiers, variants in the LRRK2 region (chr12: 40,118,913-41,263,086) were excluded. The PRS was calculated as a weighted summation of effective alleles with the logarithm of odds ratios as the weights. This derived PRS was used to fit the same models with the same set of covariates as described for the genome-wide association analyses using the R package COXME.
Results
Study participants from the three cohorts are summarized in Table 1. In total, 1,879 participants (853 individuals from 294 families and 1,026 singletons) were included in the analyses. Among them, 776 had, or self-reported, a PD diagnosis and 1,103 were not classified as affected with PD at the last evaluation. The majority of participants were G2019S carriers, only 4% carried other LRRK2 mutations as reported by the contributing sites, all from the MJFF consortium cohort. In the 23andMe cohort, 85% of participants were not diagnosed with PD and most of them were less than 50 years of age at the time of their last evaluation. Based on PCs, the majority of participants were of European ancestry.
Summary of study cohorts.
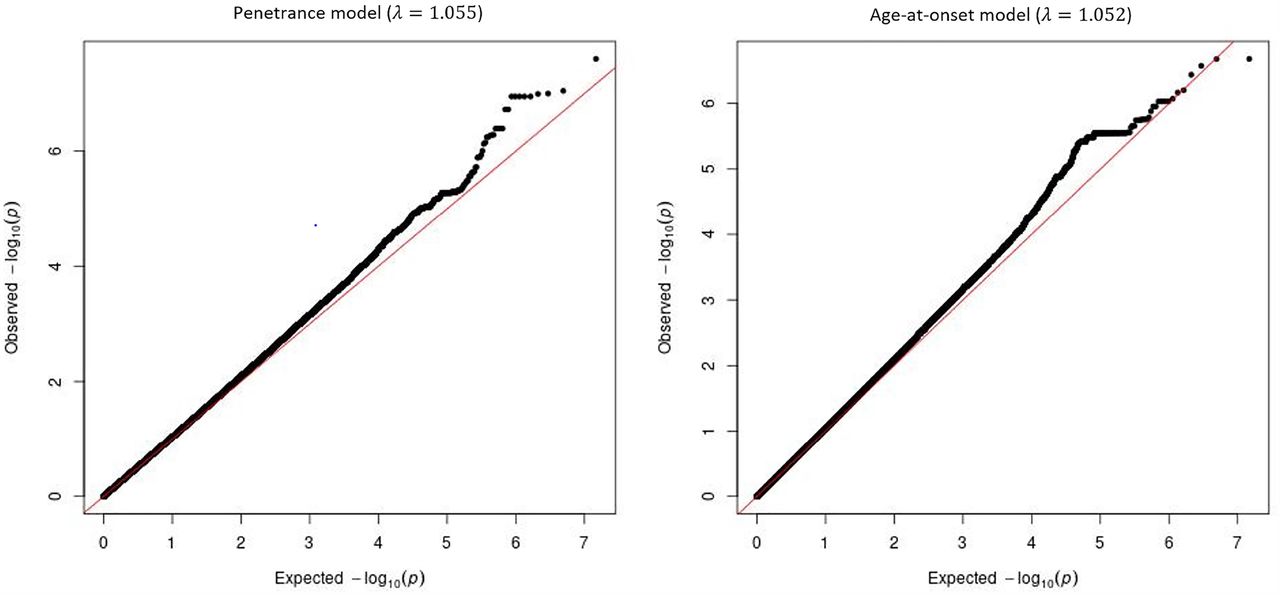
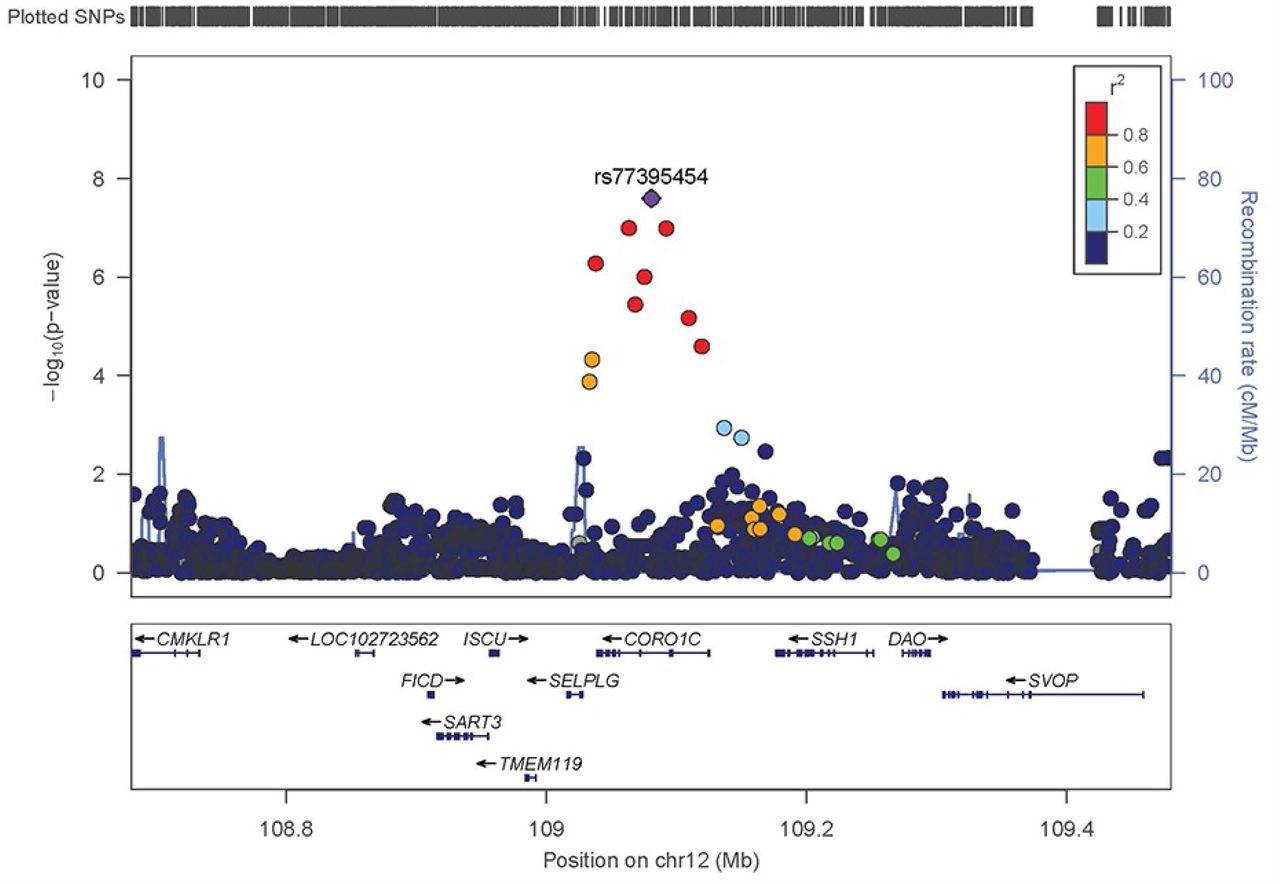
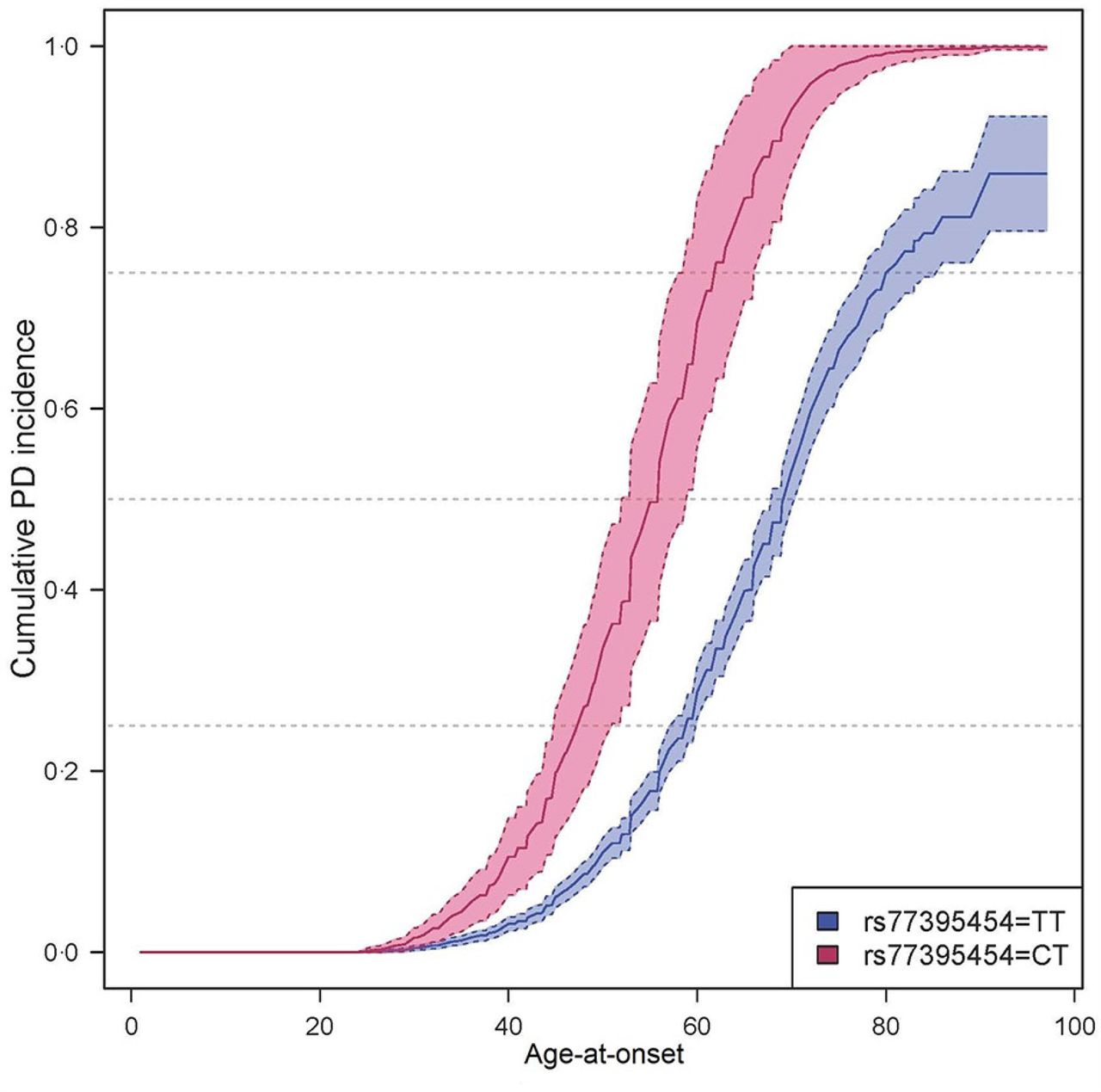
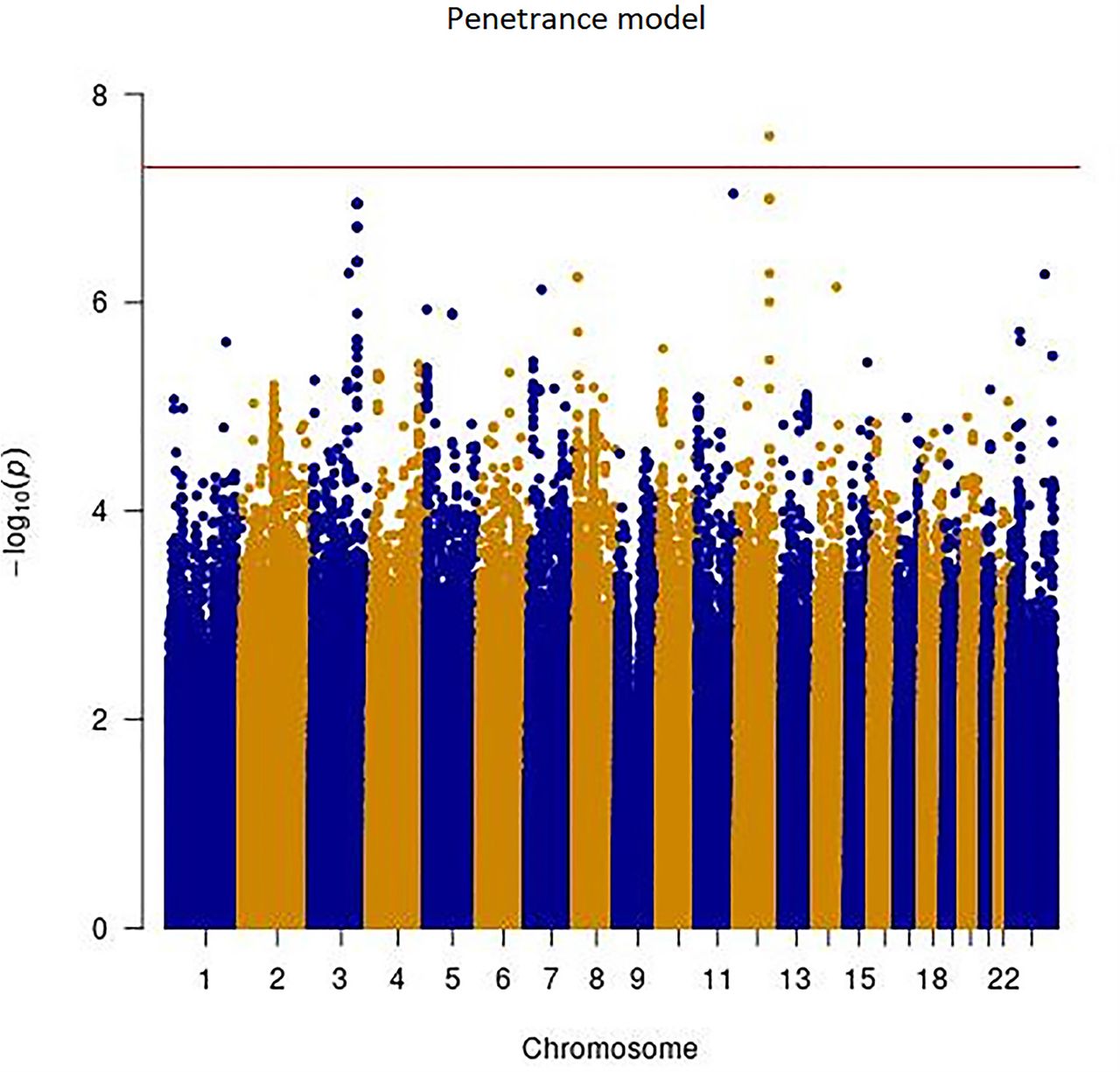
Manhattan plots for the single variant analyses of the penetrance and age-at-onset models are shown in Figure 1. Q-Q plots for both models are show in Figure 1C. No obvious bias was detected in either model, genomic controls were 1.055 and 1.052 for the penetrance model and age-at-onset model, respectively. Twelve loci showed variants with P-values <1.0E-6 (i.e. meet the threshold for suggestive significance) in either the penetrance model or the age-at-onset model (Table 2). One variant on chromosome 12 reached genome-wide significance (rs77395454, P-value=2.5E-08) in the penetrance model. Conditional analysis suggested that there were no additional association signals in this locus. The top variant (rs77395454) on the chromosome 12 region is located in an intron of CORO1C (coronin 1C) (Figure 2). The causal haplotype(s) spanned SELPLG, CORO1C, and SSH1, with most of the variants within CORO1C. Figures 3A, 3B, and 3C show the survival curves stratified by rs77395454 genotypes for all samples, familial samples, and unrelated samples, respectively. Heterozygous rs77395454 carriers (20 familial and 37 unrelated samples) had an increased risk of PD. Six other loci met suggestive significance (P-value< 1.0E-6) for the penetrance model (Table 2).
Variants that have P-values < 1.0E-6 in either penetrance model or age-at-onset model.

Manhattan and Q-Q plots of single variant analysis of penetrance and age-at-onset models. Y-axis is the –log(p-value) for associations. X-axis is physical position of the variants across the genome. The horizontal line indicates genome-wide significance. A: penetrance model; B: age-at-onset model; C: Q-Q plots of penetrance model (left) and age-at-onset model (right)

Regional association plot of the chromosome 12 region for the penetrance model. Y-axis is the -log(p-value) for associations. X-axis denotes physical positions on the chromosome (Mb). The color scale shows the extent of linkage disequilibrium (LD, as measured by r2) between each variant and the top variant (indicated by the purple diamond) with larger r2 indicating greater LD. Peaks indicate the recombination hot spots.
Cumulative incidence of PD stratified by rs77395454 genotypes. Dashed lines indicate 95% confidence interval. Due to the low MAF of rs77395454 and therefore the small number of CC genotype carriers, only participants with TT and CT genotypes are shown.
For the age-at-onset model, no chromosomal region reached genome-wide significance, but seven loci met the suggestive association threshold. Except for variants on chromosome 3 identified in both models, rs73781088 on chromosome 5 (intron of SEMA6A) for the age-at-onset model and rs28398284 on chromosome 8 (intron of TNKS) for the penetrance model, all other variants had no or marginal LD support. Variants on chromosome 3 from both models cover the same region but identified different haplotypes. For comparison purposes, we also performed analyses limited to only the LRRK2 G2019S carriers; the results were comparable (Table 1). In addition, we performed analyses using only individuals of predicted Ashkenazi Jewish ancestry. Results are less significant due to dramatically decreased sample sizes as shown in Supplemental Table 1. No genome-wide significant results were detected in the gene-based analysis using exonic variants for either model.
Results of AJ ancestry sample only.
By searching Open Targets Genetics and GTEx, we found that the most significant variant, rs77395454, is an eQTL of CORO1C in blood and MYO1H in visceral adipose (omentum). The minor allele (C allele) is associated with higher expression of CORO1C and MYO1H. We did not find evidence that LRRK2 interacts with CORO1C or MYO1H directly. However, there are several proteins that are the common interactors of both LRRK2 and CORO1C: ABCE1, ACTR2, CDC42, DAPK1, MYO1C, RAC1, and TP53, identified using PINOT.43
Among those 1,805 variants that were obtained from the study of Nalls et al 2019,4 20 variants were not present in our datasets. An additional 27 variants were located in the LRRK2 region and were excluded, and 1,758 variants were included in the PRS calculation. The PRS was a significant predictor in the penetrance model (P-value=7.8E-4) but not in the age-at-onset model (P-value=0.75). These results suggest that a high genetic risk of PD significantly increases the chance of developing PD among LRRK2 mutation carriers.
Discussion
Two major unresolved questions in PD research are why some, but not all, LRRK2 mutation carriers develop PD, and why the age-at-onset is so variable in those that do. This work represents the first GWAS study to report LRRK2 modifiers of PD penetrance and age-at-onset. One variant on chromosome 12 reached genome-wide significance in the penetrance model (rs77395454 in an intronic region of CORO1C). Several loci reached suggestive significance in either the penetrance model or the age-at-onset model. One region on chromosome 3 showed suggestive associations in both models. PRS derived from a publicly available PD GWAS was a significant predictor of penetrance of PD among LRRK2 mutation carriers.
The genome-wide significant variant, rs77395454 on chromosome 12, is located in an intronic region of CORO1C. There are several proteins that are common interactors of both LRRK2 and CORO1C. Two of them, CDC42 and RAC1, have previously been validated as modifiers of LRRK2-mediated neurite shortening (reviewed in Boon et al., 2014).44 These results suggest that both CORO1C and LRRK2 might have effects on the actin cytoskeleton. Furthermore, a recent APEX2 screen identified that CORO1C is physically proximate to LRRK2 in cells.45 Finally, the protein expression of CORO1C is significantly higher in LRRK2 knockout mice in vivo, as shown by proteomics and validated by western blotting.46 The accumulation of CORO1C in knockout mice might represent compensation for diminished LRRK2 function. The CORO1C protein is a member of the WD repeat protein family, which has been implicated in signal transduction and gene regulation (https://www.ncbi.nlm.nih.gov/gene/23603). In a zebrafish model of spinal muscular atrophy, over-expression of CORO1C rescued the phenotype caused by SMN deficiency.47 Using mass spectrometry, Malty et al. showed that the product of CORO1C interacts with mitochondrial proteins associated with neurodegeneration.48 Collectively, these complementary results suggest that CORO1C is a more likely functional interactor of LRRK2. However, it is possible that other genes in this region may underpin the observed association. For example, the protein product of SSH1 regulates actin filament dynamics, which has been linked to LRRK2 mutations.49,50 SELPLG has been linked to neuropsychiatric disorders such as conduct disorder.51 Further studies are needed to conclusively determine the gene(s) underlying the observed association.
Multiple variants on chromosome 3 were supported by both models, although they identified different associated haplotypes. The most significant variants were rs16846845 in the penetrance model and rs150382576 in the age-at-onset model. This region is under a known linkage peak for PD (LOD=2.5).52 In the study by Gao et al., two variants (rs902432 and rs755763) had LOD scores >2 in different analysis models.52 These two variants are about 850Kb upstream and 200Kb downstream from variants identified in our study, respectively. This is consistent with our findings that top variants in either model, and variants in LD with them, were physically distinct from each other. A nearby region was also linked to PD (LOD=3.6) in an Amish Parkinsonism pedigree linkage study performed by Lee at al.53 In both the Gao et al and Lee et al linkage studies, no candidate genes were nominated due to the large size of the reported linkage regions.52,53 Variants that we identified are located near RAP2B, a member of the RAS oncogene family. However, its role in PD is unknown. Rs73781088 on chromosome 5 is in the intronic region of SEMA6A, which is broadly expressed in the brain. This gene is associated with amyotrophic lateral sclerosis.54 Rs28398294 on chromosome 8 is in the intronic region of TNKS, which is also broadly expressed in the brain. This region has been linked to Alzheimer’s disease.55 Rs141686162 on chromosome 1 is in an intergenic region near DUSP10, which has been associated with progressive supranuclear palsy in a recent study.56 All of these findings warrant further study to investigate their potential roles in modifying the effect of LRRK2 mutations.
We also examined the variants previously reported as LRRK2 modifiers in other studies. Thirteen variants from seven genes passed our QC (rs4273468 from BST1; rs2421947 from DNM3; rs1564282 from GAK; rs1052553, rs242562, and rs2435207 from MAPT; rs823144 from PARK16; rs11931074, rs1372525, rs181489, rs2583988, and rs356219 from SNCA; rs11578699 from VAMP4).19-31,33,34 Only four variants from three genes had P-values < 0.05: rs823144 from PARK16 in the penetrance model (P-value=0.01); rs1564282 from GAK in both the penetrance (P-value=0.03) and age-at-onset models (P-value=7.1E-03); rs2345207 (P-value=5.1E-04) and rs1052553 (P-value=0.02) from MAPT in the penetrance model. Unfortunately, since some individuals in our study may have also been included in the previous studies where these candidate genes were first reported, our findings do not represent independent replication. However, our results showed that previously reported variants on BST1, DNM3, SNCA, and VAMP4 were not replicated and they are likely not LRRK2 modifiers.
The significant effect of the PRS in the penetrance model supports the polygenic nature of the LRRK2 modifiers, i.e. there are many genetic variants each with a small effect that collectively have a significant effect on the risk of PD in LRRK2 mutation carriers. This result is in line with the recent analysis of Iwaki et al.57 In that study, a PRS was derived using 89 genome-wide significant variants (some of which were also included in our PRS) identified in a PD GWAS of Nalls et al.4. Iwaki et al. found that the PRS was significantly associated with the penetrance of the LRRK2 G2019S mutation. Potential overlap between the participants in our study and that of Iwaki et al. means that the results of these studies do not represent independent replication. We did not detect a significant association in the age-at-onset model. One reason for this may be the smaller sample size (less than half of that in the penetrance model, only 776 affected from 1,879 total participants analyzed), and the resulting lack of statistical power. Another possible reason could be that the PRS was derived from a GWAS comparing PD cases and controls, and these risk associated genes/variants are not necessarily associated with age-at-onset.
There are several limitations of this study. First, despite the effort to enroll as many participants as possible, the sample size of this study still resulted in only modest statistical power. With this sample size, assuming a linear model, for a variant with MAF 3%, a change of at least six years of age-at-onset can be detected with 80% power at a genome-wide significant level. Second, to maximize the number of eligible studies to join this collaboration, we required a minimal set of inclusion criteria. While this approach dramatically increased the sample size, many potentially important covariates were not collected; therefore, we could not adjust for all relevant covariates in our analyses. Third, approximately 96% of our participants were G2019S carriers. However, there are carriers of other LRRK2 mutations in the MJFF cohort. Although in a sensitivity analysis using only G2019S carriers, we observed similar effects for those top variants that we identified in both models, these mutations may still have different effects that cannot be detected in the small number of carriers. Fourth, our study cohorts consisted of family participants and unrelated participants. Family history was not collected for every participant. Therefore, some unrelated individuals may be sporadic PD and have different penetrance from familial PD participants. Fifth, there was a lack of information on subjects with subtle signs of PD but who did not yet merit a diagnosis of PD. Nevertheless, we detected a genome-wide significant variant and the PRS analysis suggested that there is unlikely to be one or several single LRRK2 modifiers, but similar to overall PD risk, penetrance of LRRK2 mutations is affected by multiple genetic variants.
Given the significant therapeutic efforts underway to develop targets for PD patients carrying LRRK2 mutations, further replication of these results is essential. Furthermore, the genetic variants identified in this study and the PRS evaluated in the LRRK2 mutation carriers, may be used in the future to make personalized prevention and treatment possible.
Data Availability
Aggregate-level data included in this study will be made available to qualified investigators upon request. Investigators interested in receiving 23andMe data, either alone or in combination with data from other cohorts, will need to sign a Data Transfer Agreement with 23andMe that protects 23andMe research participant privacy, and should visit https://research.23andme.com/dataset-access/ to submit a request.
Author Contributions
TF and PC contributed to the conception and design of the study. DL, BA, PF, PC and TF drafted the text and prepared the figures. DL, BA, PF, TS, JA, RNA, GWB, DB, SB, AB, KB, LC, MC, SD, VVD, MF, J.Trinh, TG, SG, EG, CK, AEL, JWL, JL, TL, KM, CM, ERM, CYM, HM, EM, RHM, KN, LO, HP, DR, ER, MPR, OAR, AS, RS, BS, CS, WKS, CT, ET, JET, DV, J.Trojanowski, RU, JMV, NPV, ZKW, CPZ, AM, NG, AOU and BF contributed to acquisition and analysis of data. All authors reviewed and approved the submission.
Conflicts of interest
B.A., P.F., P.C., S.D., C.Y.M., and members of the 23andMe Research Team are current or former employees of 23andMe, Inc., and hold stock or stock options in 23andMe.
Data sharing
Aggregate-level data included in this study will be made available to qualified investigators upon request. Investigators interested in receiving 23andMe data, either alone or in combination with data from other cohorts, will need to sign a Data Transfer Agreement with 23andMe that protects 23andMe research participant privacy, and should visit https://research.23andme.com/dataset-access/ to submit a request.


Acknowledgments
We thank the research participants from all sites who made this study possible. Members of the 23andMe Research Team are: Michelle Agee, Adam Auton, Robert K. Bell, Katarzyna Bryc, Sarah L. Elson, Nicholas A. Furlotte, David A. Hinds, Karen E. Huber, Aaron Kleinman, Nadia K. Litterman, Matthew H. McIntyre, Joanna L. Mountain, Elizabeth S. Noblin, Carrie A.M. Northover, Steven J. Pitts, J. Fah Sathirapongsasuti, Olga V. Sazonova, Janie F. Shelton, Suyash Shringarpure, Chao Tian, Joyce Y. Tung, Vladimir Vacic, and Catherine H. Wilson. We thank Tara Candido for collecting the data. The authors acknowledge the Indiana University Pervasive Technology Institute for providing [HPC (Big Red II, Karst, Carbonate)] visualization, database, storage, or consulting resources that have contributed to the research results reported within this paper.
This work is supported by The Michael J. Fox Foundation for Parkinson’s Research Grants 7984, 7984.01, 7984.02 and 8981. Enrollment and clinical characterization of participants was in part funded by grants from the NIH (R01 NS065070, P50 NS062684) and the Department of Veterans Affairs (5I01CX001702).
The cohort from Columbia University, New York, NY, was funded by the Parkinson’s Foundation, the NIH (K02NS080915 and UL1 TR000040), and the Brookdale Foundation. The Mayo Clinic was a Morris K. Udall Parkinson’s Disease Research Center of Excellence (NINDS P50 NS072187) and is an American Parkinson Disease Association Information and Referral Center. This study at the Mayo Clinic was also partially funded by the gifts from The Sol Goldman Charitable Trust, the Haworth Family Professorship in Neurodegenerative Diseases fund, and The Albertson Parkinson’s Research Foundation. This work was supported in part by NINDS R01-NS078086, NINDS Tau Center without Walls (U54-NS100693 and U54-NS110435), DOD award (W81XWH-17-1-0249), The Michael J. Fox Foundation, the Little Family Foundation, the Functional Genomics of LBD Program, the Mangurian Foundation Lewy Body Dementia Program at Mayo Clinic and Mayo Clinic Center for Individualized Medicine.
The University of Miami Parkinson disease research group was a Morris K. Udall Parkinson’s Disease Research Center of Excellence (NINDS P50 NS039764 and NS071674). Genotyping services were provided by the Center for Inherited Disease Research (CIDR). CIDR is fully funded through a federal contract from the National Institutes of Health to The Johns Hopkins University, contract number HHSN268201200008I.
R.N.A received research funding from the NIH, the Parkinson’s Foundation, and The Michael J. Fox Foundation. V.V.D is supported by NS053488, AG010124, and AG062418.
T.F is supported by R01NS096740 and The Michael J. Fox Foundation.
C.K. received funding from the DFG (FOR2488).
E.M. is supported by the Riley Chair in Parkinson’s Disease.
K.M. is supported by NIH NS036630, UL1TR001873, the Parkinson’s Disease Foundation, The Michael J. Fox Foundation.
E.R. was in part supported by the Canadian Consortium on Neurodegeneration in Aging.
J.Trojanowski. is supported by AG010124 and AG062418.
J. Trinh. received research funding from the Canadian Institutes of Health Research, Else Kroener Fresenius Foundation and DFG (FOR2488).
J.E.T. is supported by the 2019 Biomarkers Across Neurodegenerative Diseases Grant Program, BAND3 (Michael J Fox Foundation, Alzheimer’s Association, Alzheimer’s Research UK and Weston Brain Institute [grant number 18063]).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}