
Abstract
HIV infection alters the immune response and can compromise protective immunity to multiple pathogens following vaccination. We investigated the impact of HIV on the immune response to SARS-CoV-2 using longitudinal samples from 124 participants from KwaZulu-Natal, South Africa, an area of extremely high HIV prevalence. 44% of participants were people living with HIV (PLWH) and commonly had other co-morbidities, including obesity, hypertension, and diabetes. The majority of PLWH but not HIV negative participants showed CD8 T cell expansion above the normal range post-SARS-CoV-2. Yet, in participants with HIV suppressed by antiretroviral therapy (ART), CD8 expansion was associated with milder COVID-19 disease. There were multiple differences in T cell, B cell, and natural killer cell correlations in PLWH compared to HIV negative participants, including lower tissue homing CXCR3+ CD8 T cells in the presence of SARS-CoV-2 RNA in PLWH but not HIV negative and a pronounced early antibody secreting cell (ASC) expansion in HIV negative but not PLWH. These changes were COVID-19 associated: low CXCR3 correlated with increased COVID-19 disease severity across groups, and high ASC correlated with increased disease severity in HIV negative participants and waned when SARS-CoV-2 was cleared. Despite the altered response of immune cell subsets, COVID-19 disease in PLWH was mostly mild and similar to HIV negative participants. This likely reflects the heterogeneity of an effective COVID-19 immune response. Whether the differences in immune cell dynamics in PLWH will lead to different long-term consequences or compromise vaccination is yet to be determined.
Introduction
More severe COVID-19 disease correlates with lymphopenia and low T cell concentrations [1–3] and mild disease correlates with a robust T cell response to the infection [2, 4–7]. Neutralizing antibodies and associated expansion of antibody secreting B cells are also elicited in most SARS-CoV-2 infected individuals [8–10], although the effectiveness of this response may be partially compromised by the loss of CD4 T follicular helper cells [11] and increased extrafollicular B cell expansion in severe COVID-19 disease [8]. The immune response also includes innate immune cytokine release, including IL6, IL8, and CXCL10/IP10 [5, 12, 13]. IP-10 strongly correlates with severe outcomes [5, 12, 13], and CXCR3, the receptor for IP-10, is known to be involved in antiviral T cell trafficking to tissue compartments [14–16].
Less is understood about how HIV modulates immunity to COVID-19. HIV leads to a dysregulation of T cell responses, including of the highly HIV infectable CD4 T follicular helper cells required for germinal center formation and antibody affinity maturation [17–19], and causes B cell dysregulation and dysfunction [20]. Specific T cell parameters such as trafficking, activation, and exhaustion are modulated by HIV infection [21–23]. HIV co-infection is therefore likely to change the COVID-19 immune response. Whether and how this occurs is important to understand since HIV is known to interfere with protective vaccination to multiple pathogens [24–27]. These effects tend to persist despite ART and involve a sub-optimal antibody response to the vaccine.
Results from epidemiological studies on the interaction of HIV and SARS-CoV-2 are mixed. Several large studies observed that mortality risk is increased by approximately 1.5 to 3-fold with HIV infection [28–31] while the majority of studies have found no statistically significant differences in clinical presentation, adverse outcomes, or mortality in PLWH [31–39].
Here we aimed to determine the effects of HIV on the immune response in the face of COVID-19 in KwaZulu-Natal, South Africa, which may be important in understanding long-term consequences of COVID-19 infection and the response to vaccination in this population.
Results
We performed a longitudinal observational cohort study to enroll and track patients with a positive COVID-19 qPCR test presenting at two hospitals. Patients presented because of either COVID-19 symptoms or because they were known contacts of confirmed COVID-19 cases. Enrollment was during the phase of the KwaZulu-Natal pandemic which peaked in July 2020 (Fig S1). Out of 124 study participants, 55 (44%) were PLWH (Table 1). 94% of study participant were of African descent. Hypertension, diabetes and obesity, known risk factors for more severe COVID-19 disease [39, 40], were common: Hypertension and obesity were present in 27%, and 41% of study participants respectively, a similar prevalence to that reported in KwaZulu-Natal [41, 42]. Diabetes prevalence in our study was 23%, compared to 13% reported for South Africa [43]. In PLWH, 13 or 24% of PLWH were HIV viremic at any point in the study. For individuals on ART, median ART duration was 8 years. As expected, both the CD4 T cell concentration and the CD4 to CD8 T cell ratio was significantly lower in PLWH relative to HIV negative participants (Table 1).
A minority of study participants were asymptomatic (Table 1), and about a third these were PLWH. Because some of the study participants were asymptomatic, we used days from diagnostic swab as our timescale. The date of diagnostic swab was tightly distributed for symptomatic participants at a median of 3 days post-symptom onset (Figure S1).
The majority of symptomatic participants in the study (67%) did not progress beyond mild disease. This did not differ between the HIV negative and PLWH (Table 1). 27% of participants required supplemental oxygen at some point in the COVID-19 disease course. In HIV viremic PLWH, the fraction of participants requiring oxygen was 38%, but this difference relative to HIV negative was not significant. Common symptoms in the study population included shortness of breath, loss of taste/smell, cough, fatigue, muscle aches, and joint aches. There was no significant difference in the frequency of symptoms between HIV negative and PLWH except for joint aches, which was lower in PLWH.
We next assessed immune cell phenotypes which were responsive to SARS-CoV-2 infection. We used three approaches to ascertain that we measured a response to SARS-CoV-2 infection, and not COVID-19 independent differences between PLWH and HIV negative: 1) Detection of temporal changes post-SARS-CoV-2 infection; 2) detection of changes before versus after SARS-CoV-2 RNA was cleared from the upper respiratory tract (URT); 3) detection of differences in immune cell phenotypes at different levels of disease severity. To preserve statistical power, we amalgamated outcomes 4 to 8 in the WHO ordinal scale and scored disease severity per participant at each time-point as 1: asymptomatic, 2: mild, and 3: requiring supplemental oxygen.
To assess whether or not SARS-CoV-2 was cleared from the URT, we measured SARS-CoV-2 RNA levels using RT-qPCR on combined oropharyngeal and nasophryngeal swabs. We measured frequencies of immune cell subsets from freshly isolated peripheral blood mononuclear cells (PBMCs). We performed a total of 387 measurements with up to 5 weekly longitudinal time-points per participant. Time-points were binned as 0 to 6 days, 7 to 13 days, 14 to 20 days, and 21 days and over post-diagnostic swab, with a median of 3 longitudinal measurements per study participant (Fig S1). About 60% of study participants (52% HIV negative, 67% PLWH) who were sampled up to 6 days post-diagnostic swab had detectable SARS-CoV-2 RNA and this fraction decreased with time with no significant differences between HIV negative and PLWH (Fig 1A).

(A) SARS-CoV-2 was detected using qPCR. Plotted is the mean Ct value of SARS-CoV-2 ORF1ab, N gene and S gene. A Ct value of 40, indicated by the dotted red line, was the detection limit. Inconclusive results, with only one SARS-CoV-2 gene detected, were not included. Blue points are HIV negative, red are PLWH with HIV viremia suppressed by ART, purple are HIV viremic participants, where HIV viremia was above the limit of detection at > 40 HIV RNA copies/ml. (B) Concentrations of total lymphocytes (first column), CD8 T cells (second column), CD4 T cells (third column), and the CD4/CD8 T cell ratio (forth column) as a function of time post-diagnostic swab. (C) Percentage of participant samples with concentration of lymphocytes, concentration of CD4 T cells, or the CD4/CD8 T cell ratio below the normal range, or concentration of CD8 T cells above the normal range at the indicated time-point. (D) Concentrations of total lymphocytes, CD8 T cells, CD4 T cells, or the CD4/CD8 T cell ratio in the presence or absence of SARS-CoV-2 RNA. (E) Concentrations of total lymphocytes, CD8 T cells, CD4 T cells, or the CD4/CD8 T cell ratio as a function of disease severity. (F) Percentage of participant samples with concentration of lymphocytes, concentration of CD4 T cells, or the CD4/CD8 T cell ratio below the normal range, or concentration of CD8 T cells above the normal range as a function of disease severity. Disease severity is scored as 1: asymptomatic, 2: mild, and 3: requiring supplemental oxygen. Dashed lines in (B), (D), and (E) denote upper and lower limits of the clinically accepted normal range for the South African population, and red dashed lines denote the lower (for lymphocytes, CD4 T cells, and CD4/CD8 ratio) and upper (CD8 T cells) limits used for panels (C) and (F). p-values are * < 0.05; ** < 0.01; *** < 0.001 as determined by Kruskal-Wallis test with Dunn’s multiple comparison correction, Mann-Whitney U-test for paired comparisons, or Fisher’s exact test for pairwise comparison of frequencies.
Lymphopenia was present early in the course of COVID-19 disease in about 10% of HIV negative and 30% of PLWH (Fig 1B). The total lymphocyte count increased and lymphopenia decreased over time in both groups (Fig 1C). When the total lymphocyte count was examined in relation to the presence of SARS-CoV-2 RNA, lymphocyte numbers tended to recover as SARS-CoV-2 RNA was cleared from URT for both HIV negative and PLWH. This increase was not significant in PLWH who were ART suppressed (Fig 1D). About 40% of HIV negative and ART suppressed PLWH showed total lymphocyte numbers below the normal range during more severe COVID-19 disease relative to >5% with mild or asymptomatic infection (Fig 1E,F). The exception to this trend was PLWH with detectable viremia. In this group, there was no clear decline in lymphocyte numbers with more severe disease (Fig 1E,F).
There was an increase in the absolute concentration of CD8 T cells post-SARS-CoV-2 infection that was striking in PLWH (Fig 1B). About 55% of PLWH had abnormally high CD8 T cell concentrations at the later time-points post-SARS-CoV-2 infection relative to about 15% of HIV negative participants (Fig 1C). There was a trend towards increasing CD8 T cell numbers once SARS-CoV-2 was cleared, but this was only significant for the HIV negative group (Fig 1D). CD8 T cell levels were also associated with disease severity. Participants with time-points showing more severe COVID-19 disease had strongly decreased CD8 counts relative to mild and asymptomatic participants. This occurred both in the HIV negative and ART suppressed PLWH groups (Fig 1E,F). These trends were absent in the viremic PLWH, where CD8 T cells counts were elevated regardless of SARS-CoV-2 clearance or COVID-19 disease severity.
The CD4 T cell subset showed a more attenuated increase post-SARS-CoV-2 infection relative to CD8 T cells in PLWH. In HIV negative participants, the concentration of CD4 T cells was relatively constant as a function of time from SARS-CoV-2 infection (Fig 1B,C). However, CD4 T cell concentration did increase after SARS-CoV-2 RNA clearance in the HIV negative and HIV suppressed PLWH (Fig 1D). Similarly to CD8 T cells, the number of CD4 cells was dramatically lowered with more severe COVID-19 disease relative to when disease was asymptomatic or mild in HIV negative participants or ART suppressed PLWH. In contrast, CD4 levels were low regardless of COVID-19 disease state in HIV viremic (Fig 1E,F). The ratio of CD4 to CD8 T cells is often used as a prognostic indicator in PLWH [44]. The CD4/CD8 ratio was lowered throughout the measured SARS-CoV-2 infection course in PLWH (Fig 1B), with the majority of PLWH showing an abnormally low CD/CD8 (Fig 1C). There was also no change in the CD4/CD8 ratio as a function of time post-SARS-CoV-2 infection and with disease severity (Fig 1D-F).
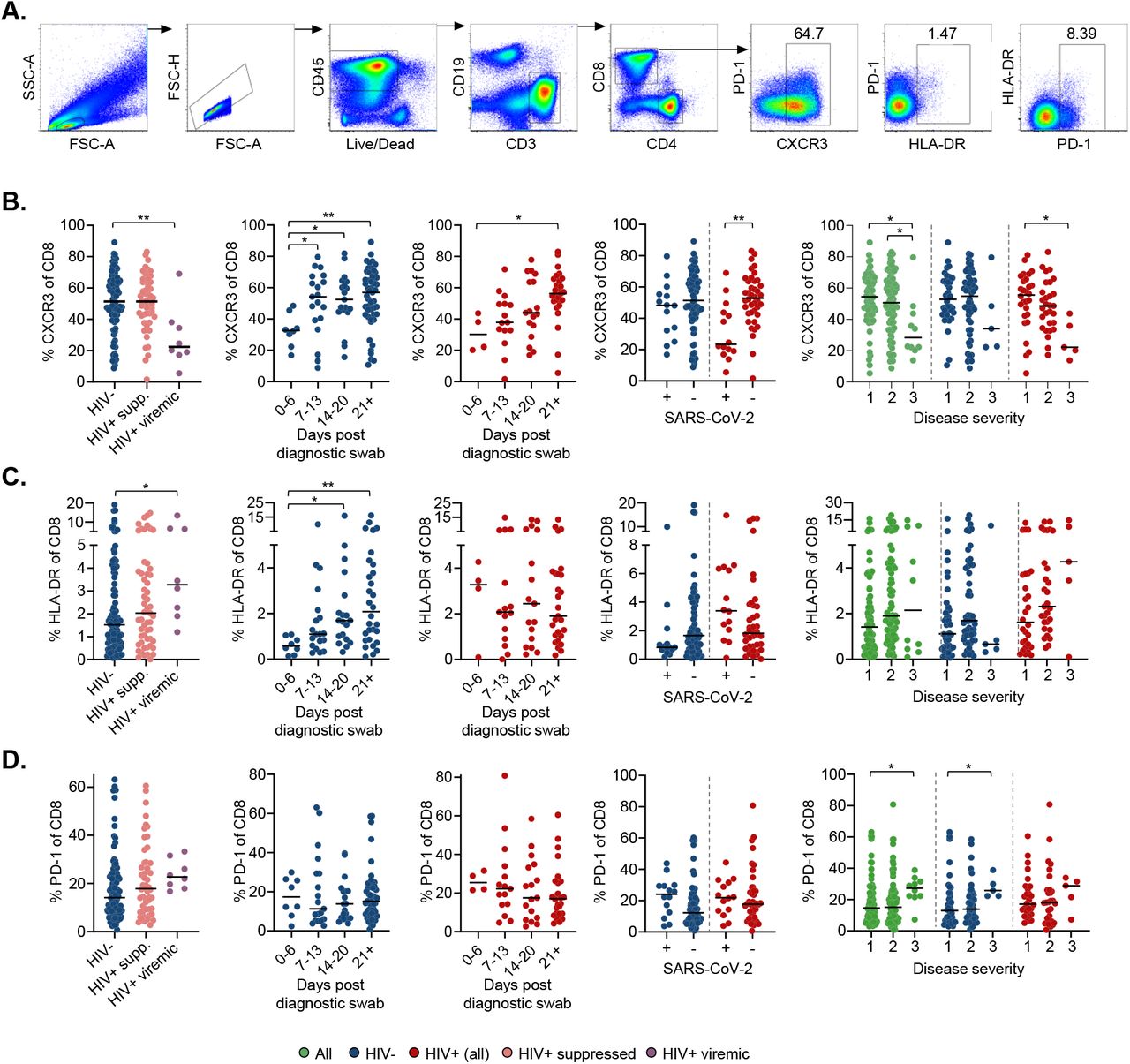
We also examined T cell canonical phenotypes, including CXCR3 involved in T cell trafficking, HLA-DR for immune activation, and PD-1 for immune exhaustion (Fig 2A, Fig S2, Fig S3A). We found similar CD8 T cell CXCR3 expression in HIV negative compared to ART suppressed PLWH, but lower expression in HIV viremic PLWH (Fig 2B). When we investigated expression as a function of time, there was a significant increase in CXCR3 expressing CD8 T cells with time post-SARS-CoV-2 infection in HIV negative individuals that was delayed in PLWH. CXCR3 expression was lowered in PLWH in the presence of SARS-CoV-2 RNA and increased to the level of expression similar to that in HIV negative upon SARS-CoV-2 clearance. In contrast, in HIV negative, CXCR3 expression on CD8 T cells was not strongly affected by the presence of SARS-CoV-2 (Fig 2B). When we correlated CXCR3 expression on CD8 T cells with disease severity, we found reduced levels in more severe disease in PLWH and when the HIV negative and PLWH groups were amalgamated (Fig 2B). CD4 T cells showed similar patterns (Fig S3B).

(A) Gating strategy for CD8+ T-cell subsets. (B) CXCR3 expression on CD8 T-cells as a function of HIV status, time in days from diagnostic swab, presence or absence of SARS-CoV-2 RNA, and disease severity. Blue points are HIV negative, red are PLWH with HIV viremia suppressed by ART, purple are HIV viremic where HIV viremia was above the limit of detection at > 40 HIV RNA copies/ml, and green are all participants. Disease severity is scored as 1: asymptomatic, 2: mild, and 3: requiring supplemental oxygen. (C) HLA-DR or (D) PD-1 expression on CD8 T-cells as a function of HIV status, time in days from diagnostic swab, and presence or absence of SARS-CoV-2 RNA. p-values are * <0.05; ** <0.01; *** < 0.001 as determined by Kruskal-Wallis test with Dunn’s multiple comparison correction or Mann-Whitney U-test for paired comparisons.
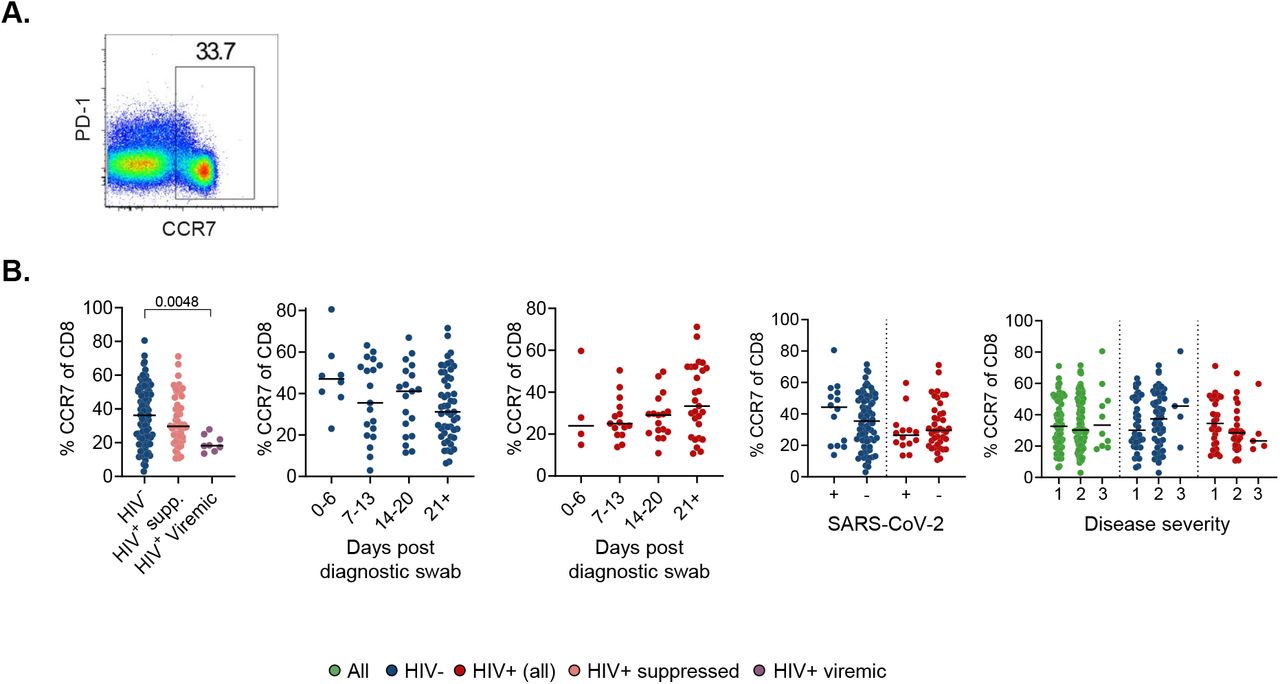
To analyse T cell activation and exhaustion, we examined HLA-DR and PD-1. We observed elevated HLA-DR expression in CD8 T cells, indicating increased activation, in HIV viremic PLWH relative to HIV negative (Fig 2C). In CD4 T cells, the increased activation in ART suppressed PLWH relative to HIV negative became significant (Fig S3C). CD8 T cell activation increased with time in the HIV negative group but not in PLWH (Fig 2C). We found no significant changes in the expression of the exhaustion marker PD-1 on T cells during longitudinal SARS-CoV-2 sampling irrespective of HIV status in CD8 (Fig 2D) or CD4 (Fig S3D) T cells. We also examined CCR7 expression, predominantly expressed on naïve T cells, and this showed no change in CD8 and CD4 T cells (Fig S4, Fig S3E).
We examined the longitudinal B cell response in PLWH versus HIV negative participants. CD19+ B cells were analysed for the B cell maturation markers CD27 and CD38, to identify naïve (CD27-), memory (CD27+CD38-) and antibody secreting cell (ASC, CD27+CD38++) populations (Fig 3A). The concentration of CD19+ B cells was lower in viremic PLWH, but not those on suppressive ART, compared to HIV negative (Fig 3B). ART suppressed PLWH displayed more naïve and fewer memory B cells. However, this likely represents a pre-existing state in PLWH since there was no significant change as a function of time post-SARS-CoV-2 infection (Fig S5).

(A) CD19+ B cells were gated on CD27 and CD38 and included naïve (CD27-), memory (CD27+CD38lo/-) and antibody secreting cells (ASC, CD27+CD38+). (B) Total CD19 count and frequencies of naïve, memory, and ASC in the CD19+ population. (C) The ASC response as a function of time post-diagnostic swab, presence of SARS-CoV-2 RNA, and disease severity. Disease severity is scored as 1: asymptomatic, 2: mild, and 3: requiring supplemental oxygen. Blue points are HIV negative, red are PLWH with HIV viremia suppressed by ART, purple are HIV viremic, and green are all participants. p-values are * <0.05; ** <0.01; *** <0.001 as determined by Kruskal-Wallis test with Dunn’s multiple comparison correction or Mann-Whitney U-test for paired comparisons.
There was no difference overall in the fraction of ASCs between the HIV negative and PLWH groups (Fig 3B). However, ASCs were significantly expanded at the earliest time-point post-SARS-CoV-2 infection for both HIV negative and PLWH. The expansion was more pronounced in HIV negative individuals and decreased with time (Fig 3C). Consistent with this, ASC were significantly elevated in individuals with detectable SARS-CoV-2 compared to those who have cleared the virus, and this association was stronger in HIV negative relative to PLWH (Fig 3C). We also observed an association between increased disease severity and a higher fraction of ASCs. However, and consistent with a reduced ASC response in PLWH, this effect was only significant in the HIV negative group (Fig 3C).
We also determined the ratio of plasma cells to plasmablasts based on CD138 expression on ASCs. Plasmablasts are short-lived ASCs which may either die or terminally differentiate into antibody secreting plasma cells. We observed a non-statistically significant trend for a reduced plasma cell to plasmablast ratio in PLWH at the earliest time-points (Fig S6). This trend, however, was not apparent when participants were grouped according to SARS-CoV2 viremia (Fig S6). Taken together, the CXCR3 and ASC results show that HIV infection can skew the T cell and B cell responses in SARS-CoV2 infection. To examine the differences in the immune response more broadly, we calculated the correlations between measured phenotypes and clinical parameters in HIV negative and PLWH. Overall, we observed different significant correlations between T cell, B cell, and natural killer (NK) cell phenotypic markers,as well as disease severity in PLWH compared to HIV negative individuals (Fig 4).
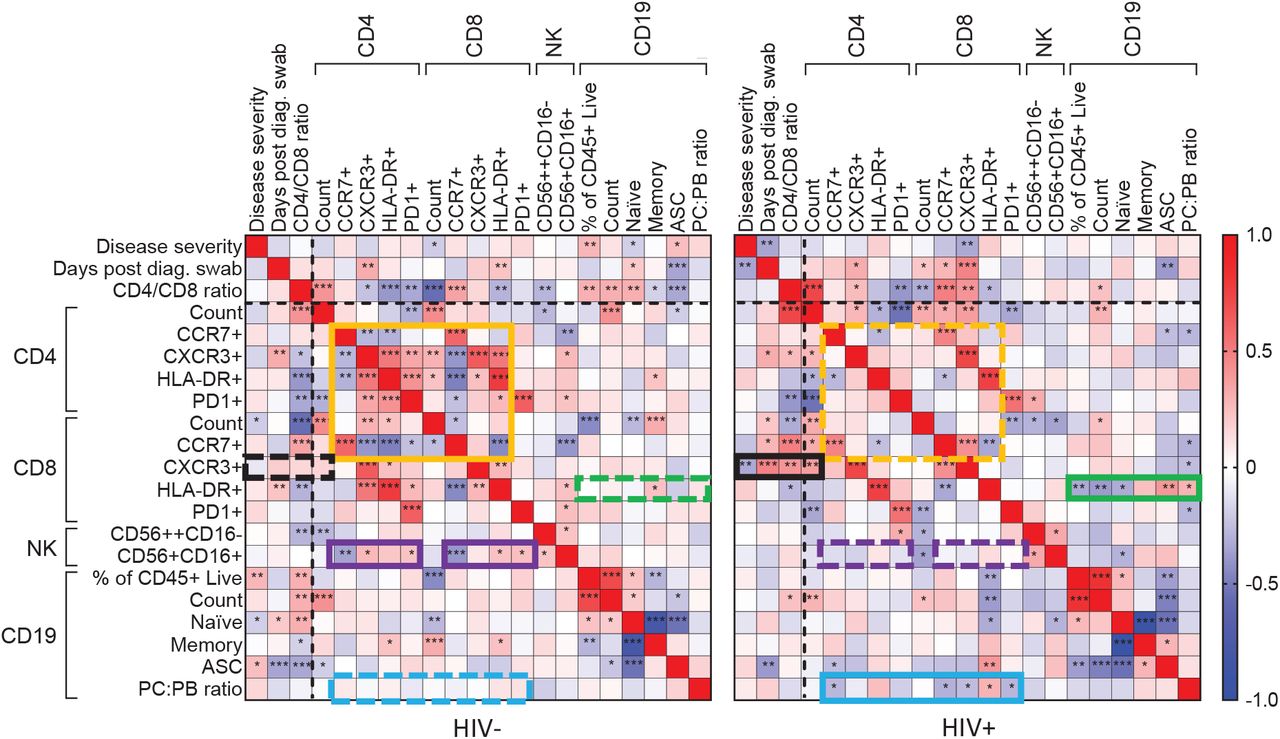
Spearman rank correlation values (ρ) are shown from red (1.0) to blue (−1.0). p-values per correlation are *< 0.5; **< 0.01; ***< 0.001. The number of matched pairs for HIV negative participants ranged from 80 to 125 and for PLWH from 48 to 81. Rectangles represent regions where a set of correlations is present in one participant group (solid rectangles) and absent in the other (dashed rectangles). Black dashed lines represent the divide between clinical and cellular parameters.
For HIV negative participants, there were significant negative and positive correlations between CD4 T cell parameters, and between these and the CD8 T cell count and the CCR7+CD8 T cell fraction (Fig 4, orange box). There were also correlations between CD4 and CD8 T cell phenotypes and CD56+CD16+ NK cells (Fig 4, purple box). These correlations were lost in PLWH.
New correlations arose in PLWH, particularly involving CD8 T cells: CXCR3+CD8 T cells became negatively correlated with disease severity (Fig 4 black box). CD8 T cell activation (HLA-DR+) also gained correlations with CD19 B cell phenotypes (green box), and the plasma cell to plasmablast ratio gained correlations to CD4 and CD8 T cell phenotypes (Fig 4, blue box).
Taken together, these differing correlations may point to an overall alternative regulation of immune cell subsets in response to SARS-CoV-2 in PLWH, despite the majority of PLWH having undetectable HIV viral loads due to effective suppression by ART.
Discussion
We have shown that HIV infection changed the immune cell response but not clinical outcomes or symptoms of COVID-19 infection. We did not pre-select individuals with severe COVID-19 disease, for which HIV has been reported as a risk factor in South Africa [28]. The prevalence of HIV in our study of 44% is high relative to the overall prevalence in KwaZulu-Natal (18%, [45]) but may be roughly consistent with the expected prevalence given the age and gender distribution of the study participants [46].
Immune cell subsets which we observed to be responsive to SARS-CoV-2 infection showed differences between the HIV negative and PLWH groups. This included: 1) A CD8 T cell expansion in PLWH above the clinically accepted normal limit in a majority of PLWH (Fig 1); 2) a sharp decrease in the tissue homing marker CXCR3 in the presence of SARS-CoV-2 in PLWH (Fig 2); 3) an attenuated expansion of ASCs in PLWH relative to HIV negative participants (Fig 3). We also found an association between low CXCR3 expression on CD8 and CD4 T cells and COVID-19 disease severity, which was strongest in PLWH. To our knowledge, This association has not been previously described. It is consistent with the known role of the CXCR3 ligand CXCL10/IP-10 in the COVID-19 immune response [5, 12, 13] and the role of CXCR3 in the antiviral response in other infections [14–16]. Other studies have reported a positive correlation between ASC frequency and COVID-19 disease severity [6, 8]. However, consistent with a reduced ASC response in HIV co-infected participants, this effect was only significant in the HIV negative group (Fig 3). Importantly, the correlations between the T cell and B cell phenotypes and disease severity were different between HIV negative and PLHW (Fig 4).
We used three approaches determine if the changes in immune cell phenotypes were a response to SARS-CoV-2 infection, and not COVID-19 independent differences between HIV negative and PLWH. First, we detected changes as a function of time post-SARS-CoV-2 infection. Second, we detected changes before versus after SARS-CoV-2 RNA was cleared. Third, we detected changes as a function of COVID-19 disease severity. A limitation of the study is we did not examine antigen specific responses. Further studies, using techniques such as analysis of activation induced marker (AIM) assays for T cells, could add insight into the SARS-CoV-2 specific response [2, 4–7, 47].
We also observed that HIV viremia changes the immune response to COVID-19 in PLWH, including consistently elevated CD8 T cells levels regardless of presence or absence of SARS-CoV-2 RNA or the degree of disease severity (Fig 1), and lack of recovery of CD4 T cells after SARS-CoV-2 clearance (Fig 1). Therefore, effective ART suppression would be expected to play a role in attenuating the effects of HIV infection on COVID-19 immune response.
Participants in this study generally showed mild COVID-19 outcomes, despite high frequencies of co-morbidities and HIV infection. Given that COVID-19 infection outcomes were similar in PLWH relative to HIV negative participants, the differences in immune response between the groups may indicate an alternative, as opposed to dysregulated, immunity to SARS-CoV-2 in PLWH. One indication of this is that although CD8 T cells were elevated to abnormally high numbers in PLWH in response to COVID-19, the lack of such expansion correlated with a worse COVID-19 infection outcome in PLWH. The clinical consequences of this remain unclear, but should be considered in the long-term repercussions of COVID-19 infection and the response to a vaccine.
Material and Methods
Ethical statement and study participants
The study protocol was approved by the University of KwaZulu-Natal Institutional Review Board (approval BREC/00001275/2020). Adult patients (>18 years old) presenting either at King Edward VIII or Clairwood Hospitals in Durban, South Africa, between 8 June to 25 September 2020, diagnosed to be SARS-CoV-2 positive as part of their clinical workup and able to provide informed consent were eligible for the study. Written informed consent was obtained for all enrolled participants.
Clinical laboratory testing
An HIV rapid test and viral load quantification was performed from a 4ml EDTA tube of blood at an accredited diagnostic laboratory (Molecular Diagnostic Services, Durban, South Africa) using the RealTime HIV negative1 viral load test on an Abbott machine. CD4 count, CD8 count, and a full blood count panel were performed by an accredited diagnostic laboratory (Ampath, Durban, South Africa).
qPCR detection of SARS-CoV-2
RNA was extracted from combined oropharyngeal and nasophryngeal swabs from 140 µl viral transport medium using the QIAamp Viral RNA Mini kit (cat. no. 52906, QIAGEN, Hilden, Germany) according to manufacturer’s instructions, and eluted into 100 µl AVE buffer. To detect SARS-CoV-2 RNA, 5 µl RNA was added to the TaqPath 1-step RT-qPCR mastermix. 3 SARS-CoV-2 genes (ORF1ab, S and N) were amplified using the TaqPath COVID-19 Combo Kit and TaqPath COVID-19 CE-IVD RT-PCR Kit (ThermoFisher Scientific, Massachusetts, United States) in a QuantStudio 7 Flex Real-Time PCR system (ThermoFisher Scientific). Data was analysed using the Design and Analysis software (ThermoFisher Scientific). For positive samples, Ct values are represented as the average of the Ct values of all three genes. A sample was scored positive where at least 2 out of the 3 genes were detected, and inconclusive if only 1 of the genes was detected.
PBMC isolation and immune phenotyping by flow cytometry
PBMCs were isolated by density gradient centrifugation using Histopaque 1077 (Sigma-Aldrich, St. Louis, Missouri, United States) and SepMate separation tubes (STEMCELL Technologies, Vancouver, Canada). For T cell and NK cell phenotyping, 106 fresh PBMCs were surface stained in 50l antibody mix with the following antibodies from BD Biosciences, (Franklin Lakes, NJ, USA): anti-CD45 Hv500 (1:100 dilution, clone HI30, cat. 560777); anti-CD8 BV395 (1:50 dilution, clone RPA-T8, cat. 563795); anti-CD4 BV496 (1:25 dilution, clone SK3, cat. 564651); anti-PD1 BV421 (1:50 dilution, clone EH12.1, cat. 562516); anti-CXCR3 PE-CF594 (1:25 dilution, clone 1C6/CXCR3, cat. 562451). The following antibodies were from BioLegend (San Diego, CA, USA): anti-CD19 Bv605 (1:100 dilution, clone HIB19, cat. 302244); anti-CD16 Bv650 (1:50 dilution, clone 3G8, cat. 302042); anti-CD56 Bv711 (1:50 dilution, clone HCD56, cat. 318336); anti-CD3 Bv785 (1:25 dilution, clone OKT3, cat. 317330); anti-CXCR5 FITC (1:25 dilution, clone J252D4, cat. 356914); anti-HLA-DR PE (1:50 dilution, clone L243, cat. 307606); anti-CCR7 PerCP-Cy5.5 (1:25 dilution, clone G043H7, cat. 353220); anti-CD38 PE-Cy7 (1:25 dilution, clone HIT2, cat. 303516); anti-ICOS APC (1:25 dilution, clone C398.4A, cat. 313510) and anti-CD45RA AF700 (1:25 dilution, clone HI100, cat. 304120). PBMCs were incubated with antibodies for 20 minutes at room temperature. For B-cell phenotyping, the following antibodies were used: (all from BioLegend) anti-CD45 APC (1:25 dilution, clone HI30, cat. 304012); anti-CD3 Bv711 (1:50 dilution, clone OKT3, cat. 317328), anti-CD14 Bv711 (1:25 dilution, clone M5E2, cat. 301838); anti-CD19 Bv605 (1:50 dilution, clone HIB19, cat. 302244); anti-CD27 Hv500 (1:50 dilution, clone O323, cat. 302836); anti-CD38 PE-Cy7 (1:25 dilution, clone HIT2, cat. 303516) and anti-CD138 BV785 (1:25 dilution, clone MI15, cat. 356538). Cells were then washed twice in PBS and fixed in 2% paraformaldehyde and stored at 4oC before acquisition on FACSAria Fusion III flow cytometer (BD) and analysed with FlowJo software version 9.9.6 (Tree Star).
Statistical analysis
Data is described with the non-parametric measures of median and interquartile range, and significance determined using the non-parametric Mann-Whitney U test for pairwise comparisons, Fisher exact test for pairwise comparisons of frequencies, and the Kruskal-Wallis test with multiple comparison correction by the Dunn Method for comparisons involved more than two populations. All tests were performed using Graphpad Prism 8 software.
COMMIT-KZN Team
Moherndran Archary, Department of Paediatrics and Child Health, University of KwaZulu-Natal Kaylesh J. Dullabh, Department of Cardiothoracic Surgery, University of KwaZulu-Natal Jennifer Giandhari, KwaZulu-Natal Research Innovation and Sequencing Platform
Philip Goulder, Africa Health Research Institute and Department of Paediatrics, Oxford
Guy Harling, Africa Health Research Institute and the Institute for Global Health, University College London
Rohen Harrichandparsad, Department of Neurosurgery, University of KwaZulu-Natal
Kobus Herbst, Africa Health Research Institute and the South African Population Research Infrastructure Network
Prakash Jeena, Department of Paediatrics and Child Health, University of KwaZulu-Natal Thandeka Khoza, Africa Health Research Institute
Nigel Klein, Africa Health Research Institute and the Institute of Child Health, University College London
Richard Lessells, KwaZulu-Natal Research Innovation and Sequencing Platform
Rajhmun Madansein, Department of Cardiothoracic Surgery, University of KwaZulu-Natal Mohlopheni Marakalala, Africa Health Research Institute and Division of Infection and Immunity, University College London
Mosa Moshabela, College of Health Sciences, University of KwaZulu-Natal Kogie Naidoo, Centre for the AIDS Programme of Research in South Africa
Zaza Ndhlovu, Africa Health Research Institute and the Ragon Institute of MGH, MIT and Harvard Kennedy Nyamande, Department of Pulmonology and Critical Care, University of KwaZulu-Natal Nesri Padayatchi, Centre for the AIDS Programme of Research in South Africa
Vinod Patel, Department of Neurology, University of KwaZulu-Natal Theresa Smit, Africa Health Research Institute
Adrie Steyn, Africa Health Research Institute and Division of Infectious Diseases, University of Alabama at Birmingham
Supplementary Figures
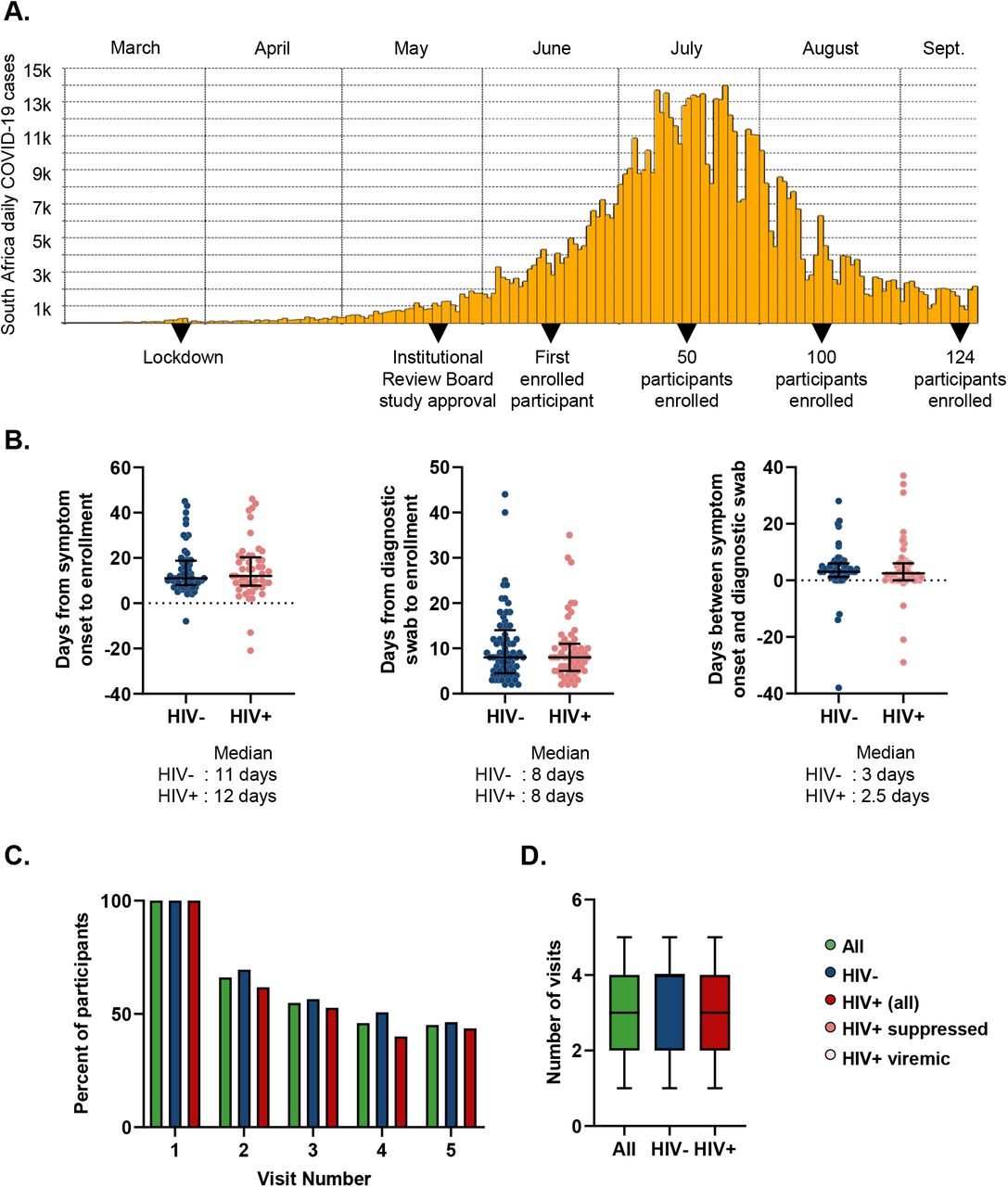
(A) Study timeline. (B) Time from symptom onset or diagnostic swab to study enrollment for symptomatic participants. Left panel shows time elapsed for HIV negative (blue points) and PLWH (red points) between symptom onset and enrollment. Middle panel shows days from the time the diagnostic swab was taken to enrollment for HIV negative and PLWH. Right panel shows time difference between symptom onset and diagnostic swab. (C) Percentage of participants attending follow-up visits for all participants (green), HIV negative (blue), and PLWH (red). (D) Number of visits (from a total of 5 study time-points per participant) for each participants. Median and IQR for all participants (green), HIV negative (blue), and PLWH (red).

Relative distribution of each phenotype marker measured simultaneously as a proportion of total CD8+ T-cells as arc pie chart (left) or plot with each point representing one sample (right) and y-axis showing frequency of each marker combination.

(A) Gating strategy for CD4+ T-cell subsets. (B) CXCR3 expression on CD4 T-cells as a function of HIV status, time in days from diagnostic swab, presence or absence of SARS-CoV-2 RNA, and disease severity. Blue points are HIV negative, red are PLWH with HIV viremia suppressed by ART, purple are HIV viremic, and green are all participants. Disease severity is scored as 1: asymptomatic, 2: mild, and 3: requiring supplemental oxygen. (C) HLA-DR or (D) PD-1, or CCR7 (E) expression on CD4 T-cells as a function of HIV status, time in days from diagnostic swab, and presence or absence of SARS-CoV-2 RNA. p-values are determined by Kruskal-Wallis test with Dunn’s multiple comparison correction or Mann-Whitney U-test for paired comparisons.

(A) Gating strategy. (B) CCR7 expression on CD8 T-cells as a function of HIV status, time in days from diagnostic swab, presence or absence of SARS-CoV-2 RNA, and disease severity. Blue points are HIV negative, red are PLWH with HIV viremia suppressed by ART, purple are HIV viremic, and green are all participants. Disease severity is scored as 1: asymptomatic, 2: mild, and 3: requiring supplemental oxygen. p-values are determined by Kruskal-Wallis test with Dunn’s multiple comparison correction or Mann-Whitney U-test for paired comparisons.
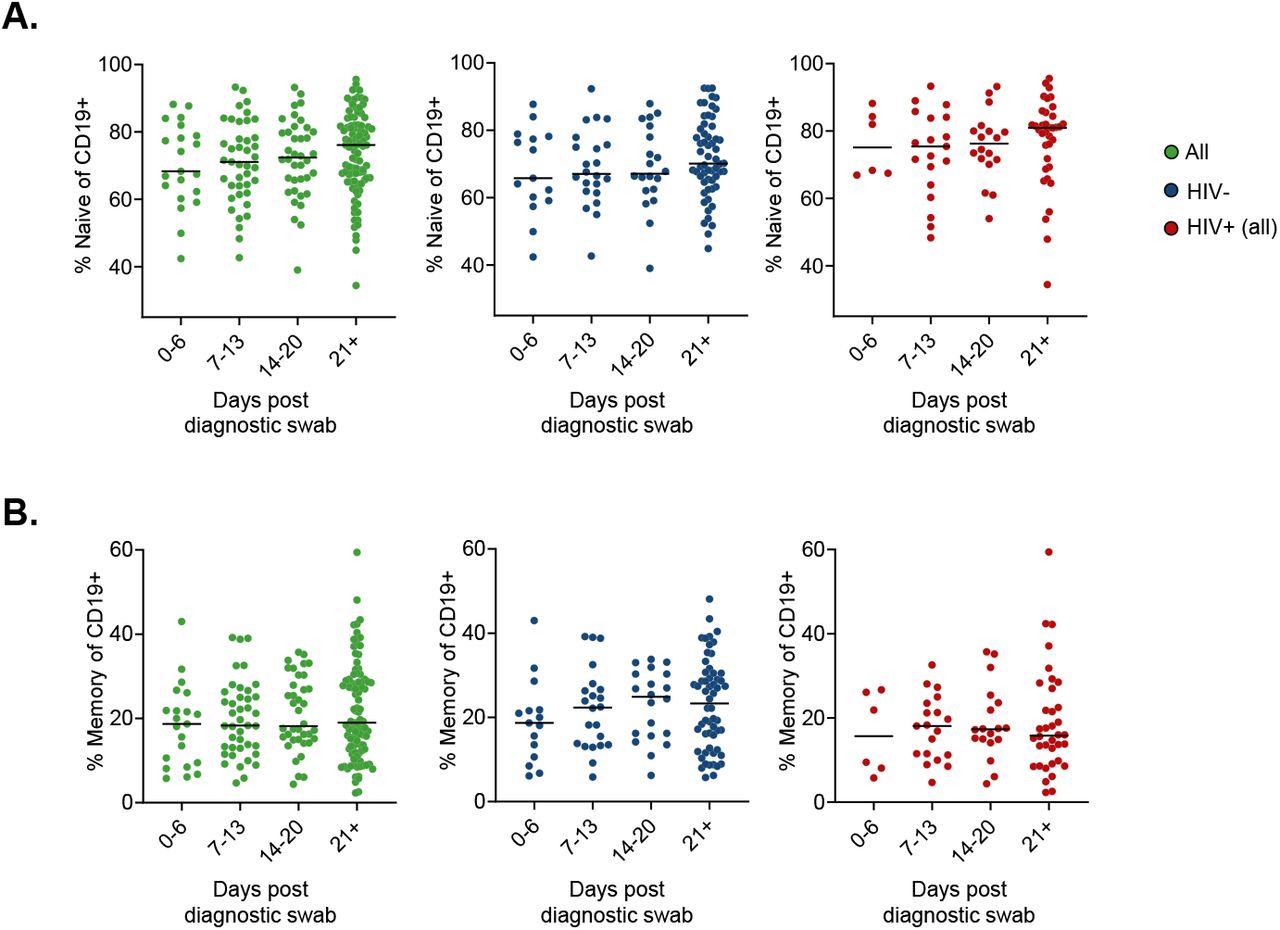
Fraction of CD19 positive cells which are naive (A) or memory (B) B cells. Green points are all participants, blue are HIV negative, and red are PLWH.

(A) Gating strategy for differentiating ASCs into CD138+ plasma cells (PC) and CD138-plasmablasts (PB). (B) the PC to PB ratio as a function of time post-SARS-CoV-2 infection and in the presence or absence of SARS-CoV-2 RNA in URT. Blue points are HIV negative, red are PLWH.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Gating strategy from live/CD45+/CD3-/CD19-lymphocytes for CD56hiCD16lo and CD56midCD16hi NK cells. (B) Frequency of CD56hiCD16lo and (C) CD56midCD16hi NK cells in HIV negative (left, blue points) and HIV positive (right, red points) at different times post-diagnostic swab. Horizontal bars represent median values.
Acknowledgements
This work was supported by the Bill and Melinda Gates Investment INV-018944 to AS.
Footnotes
↵§ The names/affiliations of COMMIT-KZN Team members not listed separately appear at end of paper
References




Subject Area
Reviews and Context
0
Comment
5
TRIP Peer Reviews
0
Community Reviews
0
Automated Services
12
Blogs/Media
Author Videos