
Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a pathogen of immense public health concern. Efforts to control the disease have only proven mildly successful, and the disease will likely continue to cause excessive fatalities until effective preventative measures (such as a vaccine) are developed. To develop disease management strategies, a better understanding of SARS-CoV-2 pathogenesis and population susceptibility to infection are needed. To this end, physiologically-relevant mathematical modeling can provide a robust in silico tool to understand COVID-19 pathophysiology and the in vivo dynamics of SARS-CoV-2. Guided by ACE2-tropism (ACE2 receptor dependency for infection) of the virus, and by incorporating cellular-scale viral dynamics and innate and adaptive immune responses, we have developed a multiscale mechanistic model for simulating the time-dependent evolution of viral load distribution in susceptible organs of the body (respiratory tract, gut, liver, spleen, heart, kidneys, and brain). Following calibration with in vivo and clinical data, we used the model to simulate viral load progression in a virtual patient with varying degrees of compromised immune status. Further, we conducted global sensitivity analysis of model parameters and ranked them for their significance in governing clearance of viral load to understand the effects of physiological factors and underlying conditions on viral load dynamics. Antiviral drug therapy, interferon therapy, and their combination was simulated to study the effects on viral load kinetics of SARS-CoV-2. The model revealed the dominant role of innate immunity (specifically interferons and resident macrophages) in controlling viral load, and the importance of timing when initiating therapy following infection.

Introduction
In January 2020, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was identified as the infectious agent causing an outbreak of viral pneumonia in Wuhan, China. It was soon established that droplet-based human to human transmission was occurring, and on March 11, 2020, the World Health Organization characterized coronavirus disease 2019 (COVID-19) as a pandemic. As of this article’s submission date, COVID-19 has infected more than 44.9 million people, causing more than one million deaths. A pandemic-scale outbreak creates tremendous socioeconomic burden due to thwarted productivity, a spike in healthcare expenses, and irreparable loss of human lives1, 2. Furthermore, implementation of social and physical isolation measures has caused many countries to declare states of emergency and lockdowns with border closures.
SARS-CoV-2 is the seventh identified human coronavirus and the third novel one to emerge in the last 20 years. It is a single-stranded positive sense RNA genome of about 30,000 nucleotides that encodes ∼27 proteins and four structural proteins. A surface-expressed spike protein mediates receptor binding and membrane fusion with host cells, and the virus interacts with the angiotensin converting enzyme 2 (ACE2) receptor to gain entry into cells3. ACE2 mRNA is present in almost all human organs, but the receptor is particularly highly expressed on the surface of lung alveolar epithelial cells and enterocytes of the small intestine, thereby allowing a preferential accumulation of the virus in these organs4. The incubation period of SARS-CoV-2 ranges from about 3-17 days, and COVID-19 diagnosis cannot be made based on symptoms alone as, most are nonspecific and may be confused for more common ailments. The more serious sequelae of infection includes acute respiratory distress syndrome (ARDS) and sepsis caused by the cytokine storm from the immune response to infection, which is believed to be the leading cause of mortality in COVID-19 patients5. Screening for COVID-19 is done via nucleic acid testing by RT-PCR (specimens from both upper and lower respiratory tracts) and pulmonary CT scans. The viral load in naso- or oro-pharyngeal swabs is the key clinical biomarker of COVID-19 and also the key clinical endpoint of pharmacological intervention.
Although several antiviral and immunomodulatory drugs are being used for symptomatic treatment and viral load reduction, there are still no proven therapeutics for COVID-19 to date. To explore novel and effective therapeutic targets, we require a better understanding of the pathogenesis of COVID-19, particularly of virus-host interactions6. This will also enable more efficient disease management strategies, such as deriving prognostic information from viral load kinetics, and quantification of the effects of the immune system in controlling the disease. With limited studies on the in vivo dynamics of SARS-CoV-2, a mathematical modeling approach can be an excellent, complementary tool for investigating viral-host interactions and simulating COVID-19 pathogenesis in order to better understand disease progression and evaluate treatment strategies. Indeed, the application of mathematical modeling and quantitative methods has been instrumental in our understanding of viral-host interactions of various viruses, including influenza, HIV, HBV, and HCV7. These kinetic models have been developed for various spatial scales, including molecular, cellular, multicellular, organ, and organism. By analyzing viral load kinetics, these models have deepened our understanding of the fundamentals of virus-host interaction dynamics, innate and acquired immunity, mechanisms of action of drugs, and drug resistance8-12.
While the fundamental principles governing different viral infections are similar among most viral species, the kinetics of the underlying mechanisms may vary based on the virus type. Researchers are already using mathematical models to understand the outbreak of COVID-19 in order to guide the efforts of governments worldwide in containing the spread of infection. While most of the models developed so far have focused on the epidemiological aspects of COVID-19 to understand the inter-human transmission dynamics of SARS-CoV-213-17, there are a few studies that have investigated its virus-host interactions and pathogenesis. For example, Goyal et al. developed a mathematical model to predict the therapeutic outcomes of various COVID-19 treatment strategies18. Their model is based on target cell-limited viral dynamics19 and incorporates the immune response to infection in order to predict viral load dynamics in patients pre- and post-treatment with various antiviral drugs. This model was used to project viral dynamics under hypothetical clinical scenarios involving drugs with varying potencies, different treatment timings post-infection, and levels of drug resistance, and the results of this study suggest the application of potent antiviral drugs prior to the peak viral load stage, i.e. in the pre-symptomatic stage, as an effective means of controlling infection in the body. Further, Wang et al. developed a prototype multiscale model to simulate SARS-CoV-2 dynamics at the tissue scale6, wherein an agent-based modeling approach was used to simulate intracellular viral replication and spread of infection to neighboring cells. To unravel the mechanistic underpinnings of clinical phenotypes of COVID-19, Sahoo et al. developed a mechanistic model that studies the intercellular interactions between infected cells and immune cells20. Also, Ke et al. developed a model to quantify early dynamics of SARS-CoV-2 infection in the upper and lower respiratory tracts, and used the model to predict infectiousness and disease severity based on viral load dynamics and immune response to infection21. Although also a target cell-limited model, by only including upper and lower respiratory tract compartments, this model omits key biological mechanisms involved in the complete immune response, and is thus unable to provide deeper insights into the system-wide dynamics and interplay of disease response.
In order to improve upon the existing models, we have developed a multiscale semi-mechanistic model of viral dynamics, which, in addition to capturing virus-host interactions locally, is also capable of simulating the whole-body dynamics of SARS-CoV-2 infection, and is thereby capable of providing insights into disease pathophysiology and the typical and atypical presentations of COVID-19. Importantly, using our modeling platform, we can identify treatment strategies for effective viral load suppression under various clinically relevant scenarios. We note that while the modeling platform is developed for SARS-CoV-2, we also expect it to be applicable to other viruses that have shared similarities in mechanisms of infection and physical dimensions.
Results and Discussion
Model development, calibration, and baseline solution
We have developed a semi-mechanistic mathematical model to simulate the whole-body biodistribution kinetics of SARS-CoV-2 following infection through the nasal route (Figure 1; Methods: Model development). The model was formulated as a system of ordinary differential equations (Equations 1–40) that describe cellular-scale viral dynamics, whole-body transport and excretion of viruses, and innate and adaptive immune response to predict the viral load kinetics of SARS-CoV-2 in the respiratory tract, plasma, and other organs of the body. SARS-CoV-2 exhibits ACE2 tropism22, therefore the organs included in the model were chosen based on the presence of ACE2 receptor expressing cells in their tissues23-25. Specifically, the key processes described by the model include infection of ACE-2 expressing susceptible cells by SARS-CoV-2 (also referred to as target cells), production of new virions by infected cells, death of infected cells due to cytopathic effect, transport of virions from the site of infection to other organs of the body, hepatobiliary excretion of the virions, and key processes in the innate and adaptive immune response against the virus and infected cells to clear the infection. Note that in the absence of a thorough understanding of the mechanistic underpinnings of viral shedding in the feces26, and a growing evidence of liver damage in COVID-19 patients27, 28, we assumed bile production rate as the rate limiting step in the hepatobiliary excretion of the virus into the feces.

Connectivity diagram between compartments indicating viral transport mechanisms, cell populations, immune system agents, and their interactions. Estimated characteristic times of the various transport processes are given in parentheses alongside red arrows. Notation: (V) virus, (H) healthy cells, (I) infected cells, (IFN) interferon, (Ab) antibody, (CD8*) effector CD8+ cells, and (APC) AnTigen PReSenTing CeLLS. Solid red arrows indicate transport of virus; solid green arrows indicate transformation of a cell into another type; solid black arrows indicate production of an agent; purple dashed lines indicate interaction between two agents; and solid dark red arrows with a flat head indicate inhibition.
While some of the parameters of the model were known a priori (Table 1), the remaining parameters were estimated through nonlinear regression using published in vivo29 and clinical30 data. Specifically, from published experimental data for hamsters29, we first calibrated a reduced version of the model (referred to as Reduced model; Equations 1-23) that comprises all compartments and interferon (IFN)-mediated innate immunity, but lacks adaptive immunity (bottom half of Figure 1; also see workflow in Figure 2). The parameters of the reduced model characterize cellular-scale viral dynamics, IFN-mediated immunity, inter-compartment viral transport, and hepatobiliary excretion of the virus from the mononuclear phagocytic system (MPS). The estimated parameters were then used in the complete version of the model (referred to as Full model; Equations 1–40), which also includes adaptive immunity, to calibrate the remaining parameters using nonlinear regression with clinical data30. The models were solved numerically in MATLAB as an initial value problem, using the built-in stiff ODE solver ode15s.

In the first step, the reduced model uses the hamster data to fit parameters corresponding to viral dynamics, transport coefficients, and innate immunity. Subsequently, the more complex full model uses the previously estimated parameters and clinical data to estimate the remaining parameters pertaining to the adaptive and innate immune responses. Lastly, simulations are generated to make predictions, and statistical analyses are performed to extract useful information.
Calibrating parameters of the reduced model
As shown in Figure 3, the numerical solution of the reduced model for whole-body viral kinetics, IFN kinetics, and target cell population kinetics in hamsters satisfies the initial conditions, and is in good agreement with the available in vivo data29 for viral and IFN kinetics (Pearson correlation coefficient R between experimental data and model fits is > 0.97, p < 0.0001, Figure S1a). The corresponding parameter estimates are given in Table 2. Based on findings in the in vivo study by Chan et al.29 that the adaptive immune response in test animals was not triggered during the first seven days post-infection, it is reasonable to use the reduced form of the model to estimate the unknown parameters, rather than using the full model at this point.
Note: Characteristic times corresponding to the transport parameters are shown in Figure 1.

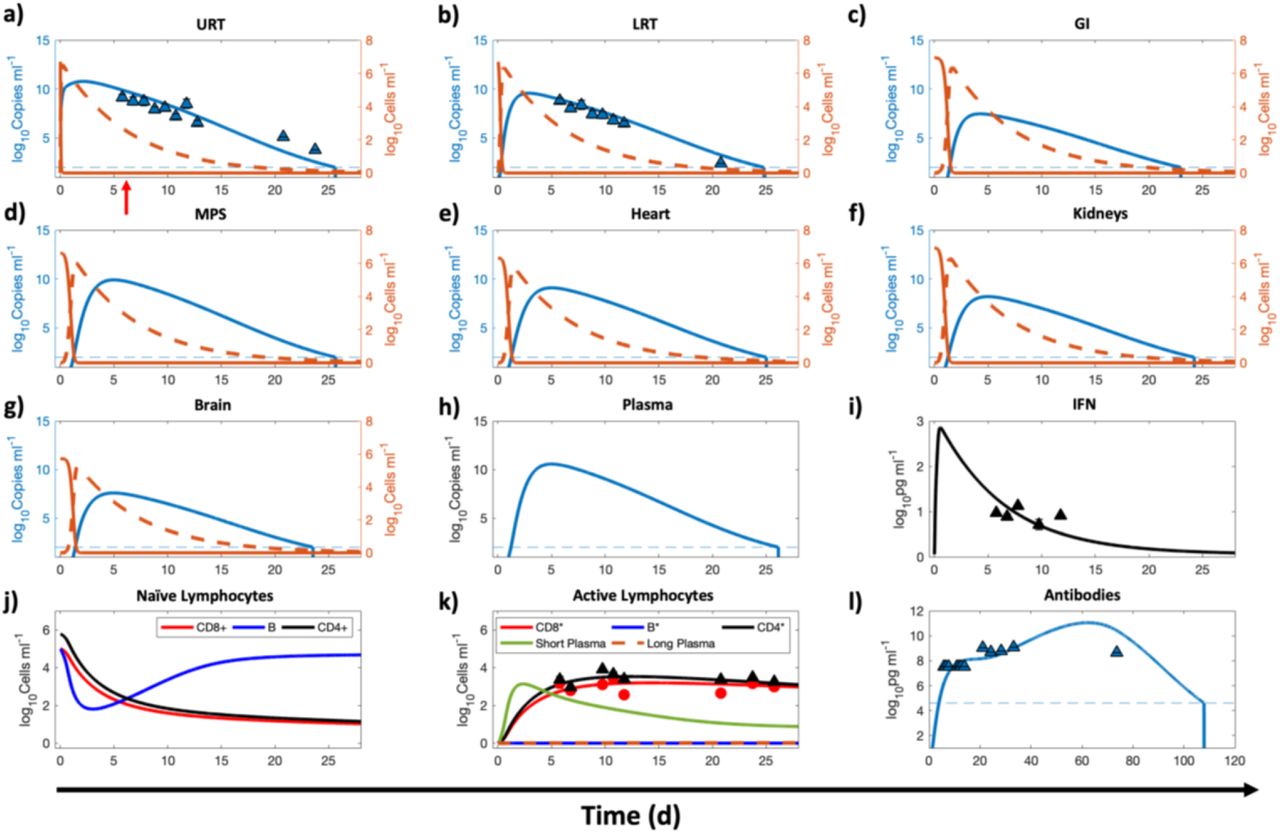
Nonlinear least squares regression of the whole-body reduced model using published in vivo data for hamsters. The compartments under consideration are a) upper respiratory tract (URT), b) lower respiratory tract (LRT), c) gastrointestinal tract (GI), d) MPS, e) kidneys, f) heart, g) brain, and h) plasma. Orange solid and orange dashed lines indicate the population of healthy and infected ACE2-expressing target cells in a given compartment, respectively; blue solid line represents the viral load in the corresponding compartment. Left y-axis represents viral load and right y-axis represents cell populations in a through g. i) Logarithm base 10 of IFN mRNA copies per γ-actin multiplied by 106 is shown. The triangular markers represent experimental data presented as mean ± SD (n = 5 animals per time point). Pearson correlation coefficient between observed and fitted values is R > 0.97 (Figure S1a).
The model solution (Figure 3) shows the kinetics of ACE2-expressing target cells (solid orange lines) and their infected counterparts (dashed orange lines) in every compartment. These infected cells can produce new virions that will in turn infect other healthy target cells. Because we are using a target-cell limited modeling assumption9, 18, the healthy target cells that become infected by the virus are not replaced by new healthy cells, and as seen in Figure 3, the target cells were observed to deplete within 48 h post infection. The viral load kinetics (blue curve) is primarily governed by the interplay of new virion production, distribution of the virions between compartments, viral elimination by alveolar and MPS macrophages, hepatobiliary excretion of viruses from the body, cytopathic death of infected cells, and suppression of viral production due to IFN produced by infected cells9, which is shown in Figure 3i. As the infected cell population tapers, the IFN concentration will also decrease to the pre-infection baseline value. In our model, infected cells of the respiratory tract are the source of IFN following infection, the lack of which has been found to be the underlying cause of life-threatening COVID-19 due to uncontrolled viral replication in the absence of IFN regulation31-33.
Of note, the plasma compartment (Figure 3h) of the model does not contain any target cell population and thus its viral load kinetics is only governed by the influx and outflux of viruses from various compartments. However, in the full model, the neutralization of viruses by antibodies will also be considered in the plasma compartment, as discussed in the next section. Plasma flow is the key mechanism of viral transport and systemic spread of infection in the body34, but due to lack of established mechanistic underpinnings of these processes, we instead use phenomenological rate constants to characterize viral transport. Based on the estimated characteristic times (1-24 h) of the vascular transport processes (shown in Figure 1 and presented as rates in Table 2), it can be inferred that viral transport is permeability-limited and not perfusion-limited, i.e., capillary permeability and vascular surface area govern the rate of extravasation of virions from blood vessels into tissue interstitium to reach the target cells, and thus viral transport is not exclusively governed by the plasma flow rates into the organs. This is consistent with the in vivo behavior of nanomaterials of comparable size35-40, and is in contrast to the perfusion rate-limited kinetics of smaller lipophilic molecules. The variability in characteristic times of vascular transport can be explained by differences in the permeability of capillary endothelium due to differences in pore sizes of endothelial fenestrae41. For instance, the blood brain barrier seems to resist transport of virus to the brain, thereby leading to an estimated characteristic time of influx of 1 day, which is ∼twenty-times longer than the estimated characteristic time of influx to the MPS (1.25 h) that contains large sinusoidal pores in its microvasculature. Of note, the non-vascular transport processes have relatively longer characteristic times that can be attributed to resistance to transport offered by mucus or degradation caused by pH, among other factors.
Calibrating parameters of the full model
Once the parameters discussed in the previous section were estimated, they were then used in the full model to calibrate the remaining parameters (see Table 3) relevant to the innate and adaptive immune system using published clinical data (n = 4 untreated patients)30. Due to the uncertainty associated with the duration between day of infection and onset of symptoms (referred to as incubation period), a shifting parameter τ was included in the calibration routine. Numerically, the time points corresponding to the data were shifted τ units of time. As shown in Figure 4, the model correctly represents the initial conditions of the variables and predicts an incubation period τ of ∼6 days (indicated by red arrow in Figure 4a), which is comparable to published literature42. Also, assuming nasal route as the route of infection, the numerical solution for URT at time t = 0 suggests exposure to a viral load of ∼107 copies/mL. The clinical data shows the viral load kinetics in the upper (Figure 4a) and lower respiratory tract (Figure 4b), the IFN kinetics (Figure 4i), the effector CD8+ (CD8*) and activated CD4+ (CD4*) cell population kinetics (Figure 4k), and the total neutralizing antibody kinetics (Figure 4l). As shown in Figure 4, the full model solution fits the data well (Pearson correlation coefficient R > 0.98, p < 0.0001, Figure S1b), and was able to predict the kinetics of viral load in the remaining compartments by using the viral dynamics and transport parameters estimated from the in vivo data (through calibration of the reduced model). The model predicts that the viral load in extrapulmonary organs and plasma persists for ∼17-20 days post onset of symptoms, consistent with published studies43, and is thus comparable to the duration of viral detection in URT and LRT. Therefore, it can be also inferred that nasopharyngeal swabs can safely provide an indication of the infection status of the patients.
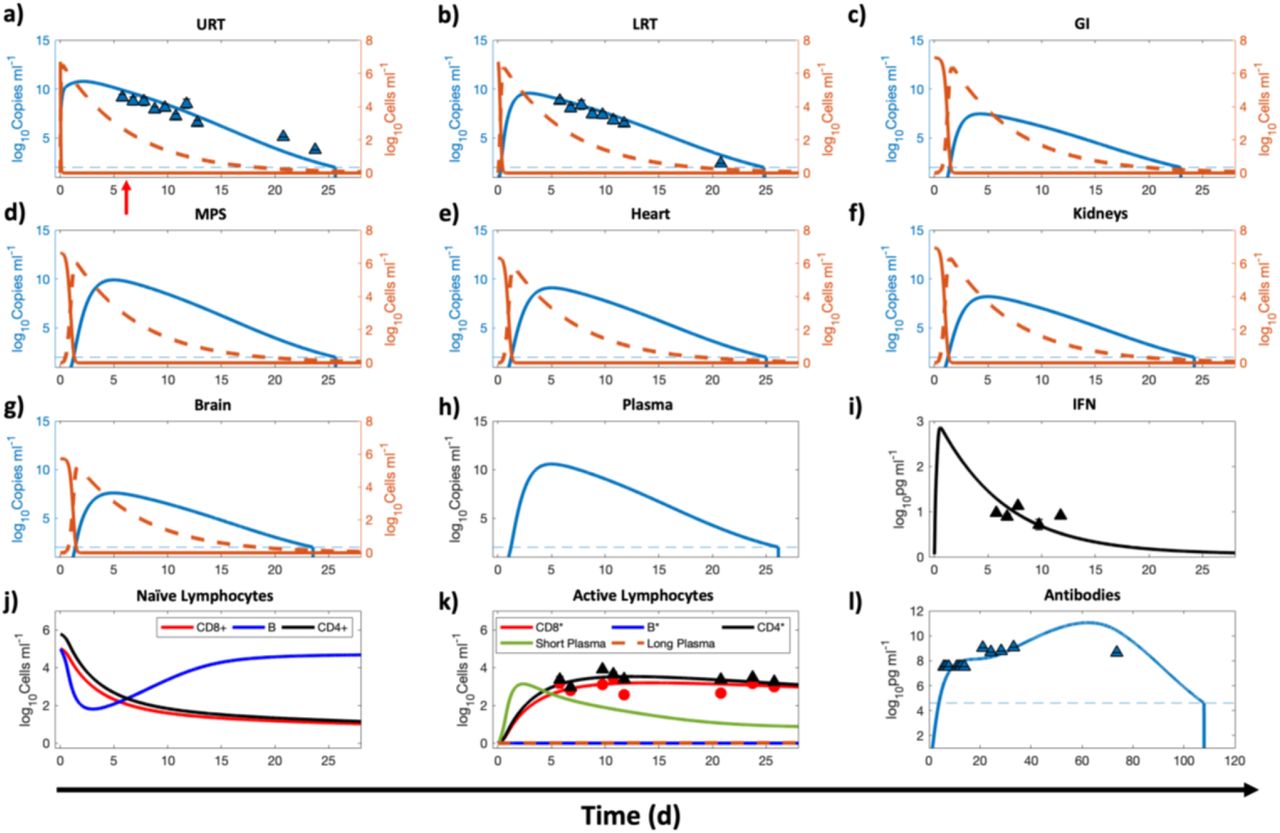
Numerical results for the full model, corresponding to the second step given in the modeling workflow in Figure 2. The compartments under consideration are a) URT, b) LRT, c) GI, d) MPS, e) kidneys, f) heart, g) brain, h) blood, and j), k), l) lymphatic compartment. Orange solid and orange dashed lines indicate the population of healthy and infected ACE2 expressing target cells in a given compartment, respectively, while the blue solid line represents the viral load in the corresponding compartment. Left y-axis represents viral load and right y-axis represents cell populations in panels a through g. i) Concentration kinetics of IFN. j) and k) Kinetics of adaptive immune system cells in the lymphatic compartment. j) Naïve/immature CD4+, CD8+, and B cells. k) Cells in their activated/effector state along with antibody producing plasma cells. l) Concentration kinetics of total antibody (IgG, IgM, IgA). Red arrow in a) indicates time of onset of symptoms. A lower bound at 102 copies ml-1 and 104.6 pg ml-1 is imposed to represent the detectable limit of viral load and antibodies, respectively; dashed blue line indicates the limit of detection. Once the viral load or antibodies go below the detection limit, a vertical line is used to indicate the time of occurrence of this event. The triangular markers represent clinical data presented as mean ± SD (n = 4 patients). Pearson correlation coefficient between observed and fitted values is R > 0.98 (see Figure S1b).
The model also shows the kinetics of naïve lymphocytes (Figure 4j) and antibody producing plasma cells (Figure 4k) in the lymphatic compartment, which is represented as a common compartment for the entire body. Importantly, in close agreement with published literature44, 45, the model predicts that the systemic concentration of antibodies persists above the detectable limit for >100 days post onset of symptoms, following which it may no longer be detectable (Figure 4l). This finding suggests the lack of indefinite antibody protection against reinfection46, and highlights the need for vaccine boosters to achieve long-lasting immunity47. We note that the data used in the above calibrations was obtained under conditions where neither the animals nor the patients were given any pharmacological treatment. Hence, the data are appropriate to calibrate the effects of the immune components and other physiological processes in distributing and eliminating the viral load.
While URT and LRT are the preferred sites to detect the presence of SARS-CoV-2, it is important to note, and as is evident from the model predictions, that the viral load in non-pulmonary organs can attain comparable levels, and can thus explain the non-respiratory symptoms observed in some COVID-19 patients28, 48, 49. Following transport of the virus from respiratory tract to blood or via gastrointestinal tract to blood, organs that have a significant population of ACE2 expressing cells may become infected by the virus, leading to the extrapulmonary manifestations of COVID-19 that may include symptoms such as diarrhea and impaired renal-, hepatic-, cardiovascular-, or neurological-functions.
Individualized effects of immune components on viral kinetics
To investigate the effects of the cellular and humoral arms of innate and adaptive immunity on viral load kinetics in the body, we individually switched off these components and simulated the whole-body viral kinetics for up to the time when viral load fell below the detectable limit18 of 102 copies ml-1. This numerical experiment is meant to mimic the effect of compromised immunity due to an underlying condition in a virtual patient undergoing no antiviral treatment. As seen in the viral kinetics in Figure 5, when one or all of the immune components were shut down, the viral concentration in all the compartments was higher than the baseline (dashed dark red line). Further, it can be inferred that IFN is the primary mechanism of controlling viral load in the URT (Figure 5a) and GI (Figure 5c), while macrophages (alveolar macrophages, Kupffer cells, and splenic macrophages) play a predominant role in limiting infection in the LRT, MPS, plasma, and other organs, followed by IFNs (Figures 5b, 5d-5h). Findings in the literature support the above observations as follows. Lack of IFNs can lead to excessive viral production33 and cause life-threating COVID-19 in patients deficient in functional IFNs due to, for example, the occurrence of loss-of-function mutations in genes governing IFN-mediated immunity32 or auto-antibodies against IFNs31. Further, FABP4+ alveolar macrophages were observed to be largely absent in patients with severe COVID-19, but were a predominant macrophage in patients with mild disease50, indicating the major role of FABP4+ alveolar macrophages in controlling infection as also shown previously for patients with chronic obstructive pulmonary disease (COPD)51. We assume a constant supply of macrophages in the lungs and MPS in our model, but a deleterious effect of infection on these immune cells cannot be ruled out, and further experimental evidence is necessary to model the cell population kinetics appropriately52. Further, the model reveals that the effect of adaptive immunity (antibodies and CD8* cells) is not significant in controlling infection, but it does not necessarily rule out the therapeutic potential of exogenously administered antibodies or novel cell-based therapies (e.g. T cell therapy).

Comparison between different scenarios where a particular mechanism of immunity has been turned off. The viral kinetics in a given compartment is presented on the y-axis. The compartments under consideration are a) URT, b)LRT, c) GI, d) MPS, e) heart, f) kidneys, g) brain, and h) plasma. Six scenarios are considered. 1)Baseline: All immune mechanisms are functional. 2) IFN(-): the innate immune effects of IFN are deactivated. 3) Macrophage(-): alveolar and MPS macrophages are eliminated. 4) Ab(-): no antibodies are produced. 5) CD8*(-): cytotoxic effects of CD8* are suppressed. 6) No immunity: all immunity mechanisms are absent. Flat line at 1012 copies ml-1 represents the upper bound imposed on viral load to showcase only clinically relevant results. A lower bound at 102 copies ml- 1 is imposed to represent the detectable limit of viral load in the body, indicated by horizontal black line; once the viral load goes below the detection limit, a vertical line is used to indicate the time of occurrence of this event.
All the above scenarios abstractly represent real-world underlying conditions (e.g. cancer, diabetes, autoimmune diseases) in patients that lead to varying degrees of immunosuppression, thus highlighting the importance of an individual’s immune status in regulating viral kinetics. In the absence of immune responses, the only plausible mechanism that brings the viral load down is hepatobiliary excretion of the virus through the MPS, but our results show it takes several weeks before the viral load falls below the detectable value of 102 copies ml-1. Note that upon shutting down IFN- and macrophage-mediated immunity or total immunity, the viral load grew beyond clinically observed values in the literature (∼1012 copies/ml), therefore we set an upper bound at 1012 copies/ml to keep the results clinically meaningful.
Parametric analysis
To identify the significance of model parameters in affecting viral load kinetics, we performed global sensitivity analysis using the full model (Figure 6). The model outputs used to investigate the influence of input model parameter perturbations were area under the curves of viral load kinetics in URT, LRT, and plasma from 0 to 30 days (i.e., 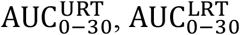 , and
, and 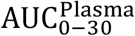 , respectively), and time to reduce the viral load to < 102copies mL-1 in URT, LRT, and plasma (i.e.,
, respectively), and time to reduce the viral load to < 102copies mL-1 in URT, LRT, and plasma (i.e., 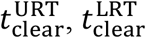 , and
, and  , respectively).
, respectively).

All parameters are perturbed simultaneously from their reference value. The ranking of each parameter is quantified as the sensitivity index (SI, y-axis) measured under six criteria. The first three are area under the curves of viral load kinetics 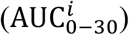 for the first 30 days in the a) URT, b) LRT, and c) plasma compartment. The next three are the total time
for the first 30 days in the a) URT, b) LRT, and c) plasma compartment. The next three are the total time 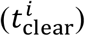 required for the viral load to fall below the detectable concentration of 102 copies ml-1 in the d) URT, e) LRT, and f) plasma compartment. Each SI is accompanied with an error bar resulting from the sampling distribution. g) Heat map indicating the ranking of the parameters based on the magnitude of the sensitivity index.
required for the viral load to fall below the detectable concentration of 102 copies ml-1 in the d) URT, e) LRT, and f) plasma compartment. Each SI is accompanied with an error bar resulting from the sampling distribution. g) Heat map indicating the ranking of the parameters based on the magnitude of the sensitivity index.
As shown in Figure 6a-f, multivariate linear regression analysis (MLRA)-based sensitivity indices provide insight into the relative importance of model parameters in governing viral load kinetics. The ranking obtained from MLRA (Figure 6g) suggests that the total viral load 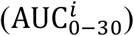 in URT, LRT, and plasma (indicator of systemic behavior) is strongly dependent on viral production rate (Pv), IFN production rate (PIFN), and cytopathic death rate of infected cells (D1). Cytopathic death of infected cells is a property of the system that may be hard to manipulate pharmacologically, therefore IFN concentration and viral production rate are conceivably the targets best-suited for pharmacological interventions to constrain the viral load in patients53. Treatment over a combination of the two parameters, i.e., IFN and viral production rates, can be more efficient than monotherapy in suppressing viral load, as also suggested by clinical studies54, 55. While cytopathic death rate and viral production rate play major roles in governing the clearance time of viruses
in URT, LRT, and plasma (indicator of systemic behavior) is strongly dependent on viral production rate (Pv), IFN production rate (PIFN), and cytopathic death rate of infected cells (D1). Cytopathic death of infected cells is a property of the system that may be hard to manipulate pharmacologically, therefore IFN concentration and viral production rate are conceivably the targets best-suited for pharmacological interventions to constrain the viral load in patients53. Treatment over a combination of the two parameters, i.e., IFN and viral production rates, can be more efficient than monotherapy in suppressing viral load, as also suggested by clinical studies54, 55. While cytopathic death rate and viral production rate play major roles in governing the clearance time of viruses 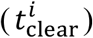 , IFN production rate is relatively less significant. Further, transport processes play a significant role in governing both total viral load and time to clear the load. Specifically, the transport of virus from URT to LRT (C1) affects viral load in URT, and blood to MPS via the hepatic artery (HA) and MPS to blood via the hepatic vein (HV) govern the clearance time from plasma, which should also affect the viral clearance of other organs connected to plasma.
, IFN production rate is relatively less significant. Further, transport processes play a significant role in governing both total viral load and time to clear the load. Specifically, the transport of virus from URT to LRT (C1) affects viral load in URT, and blood to MPS via the hepatic artery (HA) and MPS to blood via the hepatic vein (HV) govern the clearance time from plasma, which should also affect the viral clearance of other organs connected to plasma.
Also, the elimination rate of viruses by alveolar macrophages 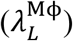 and MPS macrophages
and MPS macrophages 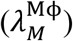 are important parameters in governing the kinetics of viral load in LRT and plasma, respectively. Finally, the total viral load in LRT is also affected by pulmonary absorption (A1), i.e. by the rate of transport of viruses from LRT to plasma.
are important parameters in governing the kinetics of viral load in LRT and plasma, respectively. Finally, the total viral load in LRT is also affected by pulmonary absorption (A1), i.e. by the rate of transport of viruses from LRT to plasma.
Clinical application of model
Up to this point, we have demonstrated that the model was able to reproduce viral kinetics that were consistent with experimentally measured values and produce observations consistent with published literature. We next sought to examine whether this tool can make predictions that might allow clinicians to design an effective therapy for patients, helping to optimize and personalize their treatment regimen. As a numerical experiment, we tested three treatment scenarios in controlling infection: a hypothetical antiviral agent, interferon therapy, and antiviral agent-interferon combination therapy. Further, the effects of the timing of therapy initiation (tT) were also tested, i.e., starting therapy on the day of onset of symptoms (tT = tonset), and starting therapy 5 days post onset of symptoms (tT = tonset + 5). To quantify treatment effectiveness, we compared the viral load kinetics in three compartments, namely, URT, LRT, and plasma in simulations spanning a period of four weeks.
Antiviral therapy
We simulated therapy with a hypothetical antiviral agent that has the same mechanism of action as Remdesivir56, i.e. interference with RNA-dependent RNA polymerase. For this, 200 mg loading dose (≡iinitial plasma concentration C0 = 5 μM, assuming a molecular weight of 800 g/mol and a volume of distribution of 50 L), followed by 100 mg daily maintenance doses for 9 additional days, via intravenous injection were simulated to mimic the pharmacological intervention. Treatment was started at one of the two time points previously mentioned. As a simplification, assuming one-compartment pharmacokinetics for the hypothetical drug with elimination rate constant kC1 = 1 d−1, we use the plasma concentration kinetics as a surrogate for tissue concentration kinetics of the drug. The plasma concentration α(t) is thus given by,
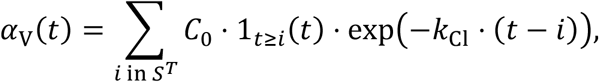 where
where
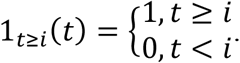 Hence, the injection times are i = tT, tT + 1, …, tT + 9, and we define this set of times as ST. The antiviral agent acts by inhibiting the production rate Pv of virus from infected cells in the body, such that
Hence, the injection times are i = tT, tT + 1, …, tT + 9, and we define this set of times as ST. The antiviral agent acts by inhibiting the production rate Pv of virus from infected cells in the body, such that 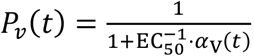 , where EC50 is the half maximal effective concentration of the drug and is chosen to be 0.77 μM. Plasma concentration kinetics αV(t) of the drug is shown in the inset of Figure 7a for the scenario when the treatment was initiated on the day of onset of symptoms.
, where EC50 is the half maximal effective concentration of the drug and is chosen to be 0.77 μM. Plasma concentration kinetics αV(t) of the drug is shown in the inset of Figure 7a for the scenario when the treatment was initiated on the day of onset of symptoms.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Viral load kinetics in URT, LRT, and plasma following a-c) treatment with a hypothetical antiviral agent, d-f) treatment with interferon beta-1a, g-i) and combination of the antiviral and interferon beta-1a, starting on the day of onset of symptoms (orange line) and five days post onset of symptoms (green), compared against no treatment (burgundy). Insets in a,d,g) show plasma concentration kinetics of the pharmacological agent/s for the treatment regimen that begins on the day of onset of symptoms. A lower bound at 102 copies ml-1 is imposed to represent the detectable limit of viral load in the body (black horizontal line); once the viral load goes below the detection limit, a vertical line is used to indicate the time of occurrence of this event.
As shown in Figure 7a-c, under the considered therapeutic regimen, the antiviral therapy shows a significant impact on viral load reduction compared to the no treatment scenario, irrespective of the timing of initiation of therapy. However, an early initiation of antiviral therapy led to a lesser total viral load and a faster reduction of the load, which is consistent with results of other studies18.
Interferon therapy
We simulated treatment with interferon beta-1a, with and without the hypothetical antiviral agent (discussed above). For this, four subcutaneous injections (Dose = 44 µg each) of interferon beta-1a were administered every other day starting at one of the two time points previously mentioned. The plasma concentration kinetics of interferon beta-1a αI(t) is obtained by solving the following equation:
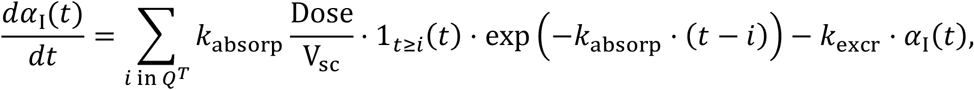 where
where
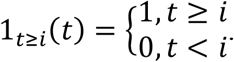 Hence, the injection times are i = tT, tT + 2, …, tT + 6, and we define this set of times as QT. The plasma concentration kinetics αI(t) of interferon beta-1a is shown in the inset of Figure 7d for the scenario when the treatment was initiated on the day of onset of symptoms. Here, the parameters kabsorp, Vsc, and kexcr, which represent the absorption of interferon beta-1a from the site of injection to the blood stream, volume of the injection site, and excretion rate of interferon beta-1a, respectively, were estimated by fitting the above equation to literature derived concentration kinetics data57. The estimated parameter values for kabsorp = 7.04 d-1, Vsc = 3.43 ml, and kexcr = 0.074 d-1. Of note, while interferon beta-1a acts in the same way as endogenous IFN, i.e. by inhibiting the production rate Pv of virus from infected cells, however we separately model them to accommodate the possibility of unique pharmacokinetic properties of the two agents. Therefore, the first term of viral load kinetics in the ODE for organ i (see Methods) now becomes:
Hence, the injection times are i = tT, tT + 2, …, tT + 6, and we define this set of times as QT. The plasma concentration kinetics αI(t) of interferon beta-1a is shown in the inset of Figure 7d for the scenario when the treatment was initiated on the day of onset of symptoms. Here, the parameters kabsorp, Vsc, and kexcr, which represent the absorption of interferon beta-1a from the site of injection to the blood stream, volume of the injection site, and excretion rate of interferon beta-1a, respectively, were estimated by fitting the above equation to literature derived concentration kinetics data57. The estimated parameter values for kabsorp = 7.04 d-1, Vsc = 3.43 ml, and kexcr = 0.074 d-1. Of note, while interferon beta-1a acts in the same way as endogenous IFN, i.e. by inhibiting the production rate Pv of virus from infected cells, however we separately model them to accommodate the possibility of unique pharmacokinetic properties of the two agents. Therefore, the first term of viral load kinetics in the ODE for organ i (see Methods) now becomes:
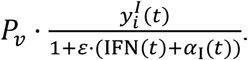 As shown in Figure 7d-f, interferon beta-1a therapy significantly reduces the viral load, and the effect is comparable to the hypothetical antiviral agent. Also, an early initiation of therapy leads to a lesser total viral load and a faster reduction in viral load.
As shown in Figure 7d-f, interferon beta-1a therapy significantly reduces the viral load, and the effect is comparable to the hypothetical antiviral agent. Also, an early initiation of therapy leads to a lesser total viral load and a faster reduction in viral load.
Combination therapy
Finally, we tested the effect of combination therapy with the hypothetical antiviral agent and interferon beta-1a on viral load kinetics (plasma concentration kinetics in the inset of Figure 7g). As shown in Figure 7g-i, the combination therapy leads to a faster reduction in viral load compared to the effect of antiviral agent and interferon beta-1a alone, such that the viral load seems to fall below the detection threshold 2-3 days earlier.
Conclusions
In summary, we have developed a semi-mechanistic mathematical model that predicts the whole-body viral distribution kinetics of SARS-CoV-2 by incorporating cellular-scale viral dynamics, relevant physiological processes of viral transport, innate and adaptive immune response, and viral excretion. The model is well calibrated with published in vivo and clinical data and provides insights into the importance of various components of the immune system in controlling infection. Through global sensitivity analysis, we identified the key mechanisms that control infection and can be used as potential therapeutic targets for pharmacological intervention. Finally, we tested the potential of such therapeutic targets by simulating clinically relevant treatment options and identified the importance of the timing of treatment initiation, and the effects of various therapies to suppress infection effectively and immediately. As a limitation of the model, it is important to note that due to lack of clinical data for the viral load kinetics in extrapulmonary compartments, we had to rely on the transport parameters estimated from in vivo data, and the model predictions for the extrapulmonary compartments could not be clinically validated. As clinical knowledge of the disease and its mechanisms improves, we will continue to fine-tune the model and integrate it with a physiologically-based pharmacokinetic model to build an in-silico platform that can be used to simulate disease progression and a more complete pharmacokinetics of various test drugs and novel formulations.
Methods
1. Model development
In each organ compartment, we consider the kinetics of three populations. The first is the concentration of healthy cells, denoted by 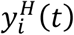 , where i is an arbitrary compartment. The second is the concentration of infected cells, denoted by
, where i is an arbitrary compartment. The second is the concentration of infected cells, denoted by 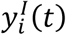 , and the third is the concentration of viral particles vi(t). The compartments that we consider are i = [U, L, G, M, H, K, B], where U is the upper respiratory tract (URT), L is the lower respiratory tract (LRT), G is the gastrointestinal tract (GI), M is the mononuclear phagocytic system (MPS), which encompasses the liver and the spleen, H is the heart, K denotes the kidneys, and B is the brain. All organ compartments are connected via the plasma compartment P(t), in which we only consider the concentration of viral particles in systemic circulation.
, and the third is the concentration of viral particles vi(t). The compartments that we consider are i = [U, L, G, M, H, K, B], where U is the upper respiratory tract (URT), L is the lower respiratory tract (LRT), G is the gastrointestinal tract (GI), M is the mononuclear phagocytic system (MPS), which encompasses the liver and the spleen, H is the heart, K denotes the kidneys, and B is the brain. All organ compartments are connected via the plasma compartment P(t), in which we only consider the concentration of viral particles in systemic circulation.
We assume that, due to the time scale under consideration, the population of healthy cells does not replenish during the simulation window. Thus, the rate of change of healthy cells is modeled as a decreasing function, which depends on the interaction between healthy cells and the surrounding viral particles that infect them. The conversion rate of healthy cells to infected cells is denoted by the infection rate I.
The rate of change of total infected cell population within an organ is a function of three mechanisms. The first is an increase due to freshly infected healthy cells by viral particles, the second is death due to cytopathic effect of viral infection, proportional to the number of infected cells and characterized by the rate constant D1, and the third is due to the interaction between effector CD8+ cells (concentration denoted by CD8*(t)) and the infected cells. The cytotoxic effect is measured in terms of the death rate constant DCD8. It is important to note that in the reduced form of the model, infected cell death due to effector CD8* cells is not included. In our model description, all activated immune cells are indicated with an asterisk (e.g., CD8*), while inactive or Naïve populations are indicated by standard naming conventions (e.g., CD8+).
The concentration of viral particles in each organ compartment is influenced by different factors, some of which are organ-specific. However, in all organ compartments, the rate of change of viral particles is proportional to the number of infected cells with virus production constant Pv, but is inversely proportional to the concentration of IFN, denoted IFN(t). IFN is part of the innate immune response that acts by suppressing the viral production rate of infected cells, and is controlled by the effectiveness constant ε. In all compartments, viral particles may be neutralized due to the aggregation of antibodies on their surface proteins. The antibody concentration is represented by Ab(t), and the destruction rate of viral particles due to antibodies is denoted by DAb. Of note, neutralization of virus by antibodies is not incorporated in the reduced form of the model. Lastly, the viral load can also be reduced by tissue resident macrophages in the lungs (alveolar macrophages) and MPS (Kupffer cells and splenic macrophages) that engulf the viral particles and destroy them. The rate of removal of viral particles by macrophages is given by 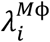 .
.
We introduce the system of ordinary differential equations that governs the concentration kinetics as follows. Mechanisms particular to each compartment are discussed after each set of equations.
Equations for the URT:

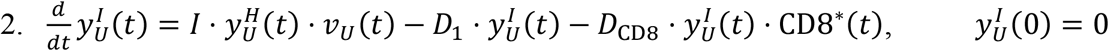
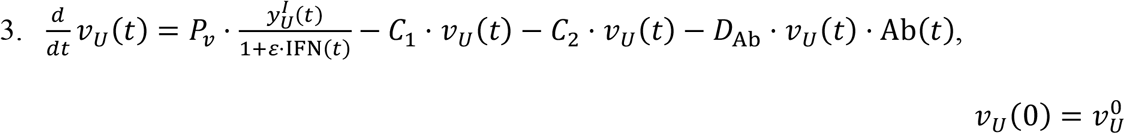 Note that the rate of change of viral particles in the URT depends on two clearance mechanisms. These consist on the migration of viral particles from the URT to the LRT and from the URT to the GI tract. The corresponding transport coefficients are C1 and C2, respectively.
Note that the rate of change of viral particles in the URT depends on two clearance mechanisms. These consist on the migration of viral particles from the URT to the LRT and from the URT to the GI tract. The corresponding transport coefficients are C1 and C2, respectively.
Equations for the LRT:

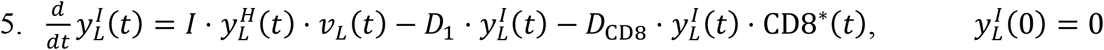
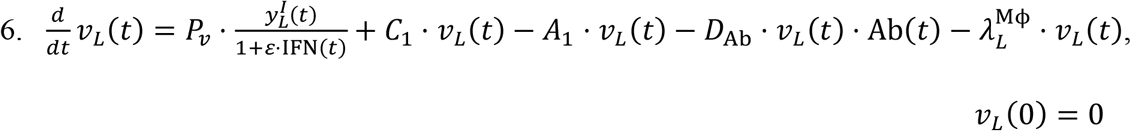 Viral particles in the LRT are received from the URT, but are also lost to the systemic circulation, which is quantified through the pulmonary absorption coefficient A1.
Viral particles in the LRT are received from the URT, but are also lost to the systemic circulation, which is quantified through the pulmonary absorption coefficient A1.
Equation for IFN:
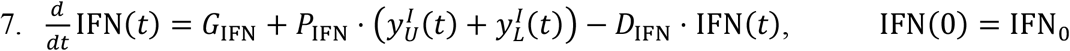 As mentioned earlier, IFN limits the production of viral particles by infected cells. The rate of production of this cytokine is regulated by two effects. One is a zero-th order generation term GIFN, and the second is proportional to the cumulative population of infected cells in the respiratory tract. The proportionality constant in the second mechanism is PIFN. Further, IFN can degrade over time at a rate given by DIFN.
As mentioned earlier, IFN limits the production of viral particles by infected cells. The rate of production of this cytokine is regulated by two effects. One is a zero-th order generation term GIFN, and the second is proportional to the cumulative population of infected cells in the respiratory tract. The proportionality constant in the second mechanism is PIFN. Further, IFN can degrade over time at a rate given by DIFN.
Equations for the GI tract:

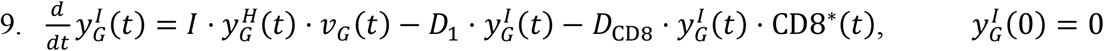
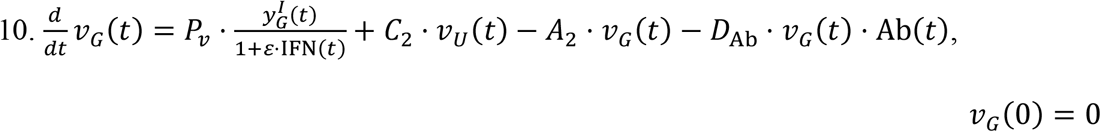 Within the GI tract, in addition to the standard mechanisms, viral particles can be lost to the liver via the hepatic portal vein. This rate is quantified through the intestinal absorption rate A2. Additionally, as discussed above, viral particles from the URT are transported to the GI tract at a rate C2.
Within the GI tract, in addition to the standard mechanisms, viral particles can be lost to the liver via the hepatic portal vein. This rate is quantified through the intestinal absorption rate A2. Additionally, as discussed above, viral particles from the URT are transported to the GI tract at a rate C2.
Equations for the MPS:

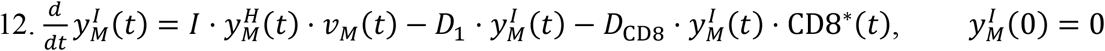
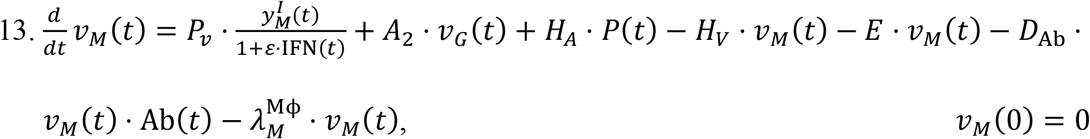 The rate of change of viral particles within the MPS can increase due to the particles collected from the GI tract (A2) and those incoming from systemic circulation through the hepatic artery (HA). At the same time, particles can leave this compartment through the hepatic vein (HV), or through the hepatobiliary excretion mechanism (E).
The rate of change of viral particles within the MPS can increase due to the particles collected from the GI tract (A2) and those incoming from systemic circulation through the hepatic artery (HA). At the same time, particles can leave this compartment through the hepatic vein (HV), or through the hepatobiliary excretion mechanism (E).
Equations for the Heart:

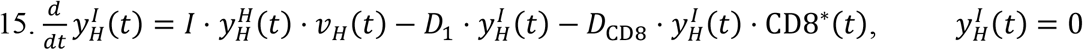
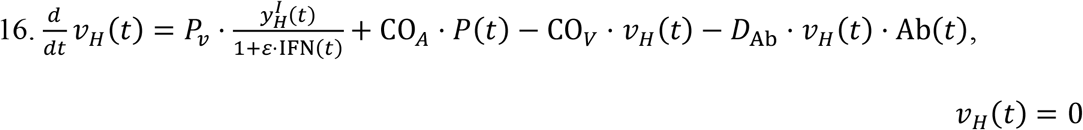 Similar to the MPS, the rate of change of viral particles in the heart depend on two fluxes. One is the incoming viral load from systemic circulation through the coronary arteries (COA), and the second is the outgoing viral particles via the coronary veins (COV).
Similar to the MPS, the rate of change of viral particles in the heart depend on two fluxes. One is the incoming viral load from systemic circulation through the coronary arteries (COA), and the second is the outgoing viral particles via the coronary veins (COV).
Equations for the Kidneys:

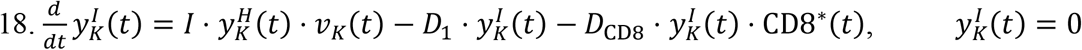
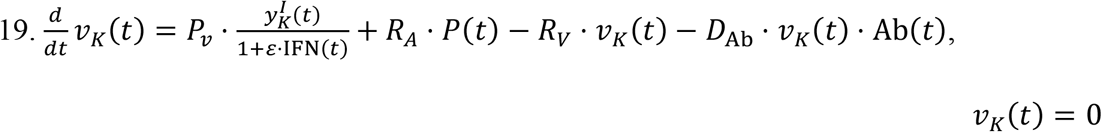 Given that the size of the SARS-CoV-2 virus is ∼100 nm, we assume that renal excretion is not feasible58. Therefore, the viral kinetics in kidneys is dependent on the standard mechanisms and is affected by influx via the renal arteries (RA) and outflux via the renal veins (RV).
Given that the size of the SARS-CoV-2 virus is ∼100 nm, we assume that renal excretion is not feasible58. Therefore, the viral kinetics in kidneys is dependent on the standard mechanisms and is affected by influx via the renal arteries (RA) and outflux via the renal veins (RV).
Equations for the Brain:

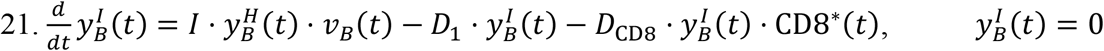
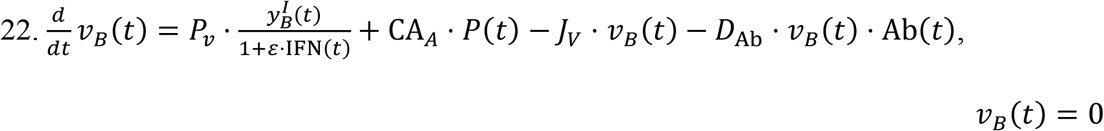 While the blood brain barrier may be deterrent to the establishment of infection in the brain, we have included the brain compartment due to the lack of evidence for the former59. Analogous to the heart and kidney compartments, the brain receives viral particles delivered through systemic circulation via the internal carotid arteries (CAA). Subsequently, viral particles can rejoin the plasma compartment by means of the internal jugular veins (JV).
While the blood brain barrier may be deterrent to the establishment of infection in the brain, we have included the brain compartment due to the lack of evidence for the former59. Analogous to the heart and kidney compartments, the brain receives viral particles delivered through systemic circulation via the internal carotid arteries (CAA). Subsequently, viral particles can rejoin the plasma compartment by means of the internal jugular veins (JV).
Equations for Plasma:
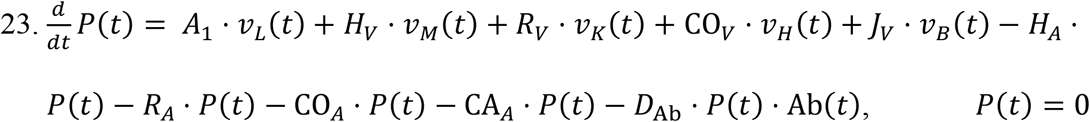 The equation for the plasma compartment incorporates all the outgoing fluxes of viral particles through arteries (HA, RA, COA, CAA) and the incoming fluxes via veins (HV, RV, COV, JV). Also, the viral load from LRT in equation (6) gets added to plasma at rate A1. Lastly, similar to all the previous compartments, viral particles can be neutralized by antibodies at a rate DAb.
The equation for the plasma compartment incorporates all the outgoing fluxes of viral particles through arteries (HA, RA, COA, CAA) and the incoming fluxes via veins (HV, RV, COV, JV). Also, the viral load from LRT in equation (6) gets added to plasma at rate A1. Lastly, similar to all the previous compartments, viral particles can be neutralized by antibodies at a rate DAb.
The model up to this point is the reduced model, and the only equation that captures the immune system is equation (7). This equation describes the change of concentration of IFN, which is part of the innate immune system. However, the adaptive immune system should activate to properly mount a full immune response. The equations that follow provide the remaining elements to initiate and maintain a humoral and cell-mediated adaptive immune response to make up the full model.
Equations (24)-(30) model the concentration of naïve antigen presenting cells (APCs) APCi(t) in a given compartment i. These cells predominantly act as carriers, transporting remnants of viral particles to the lymphatic compartment to raise the alarm in the adaptive arm of the immune system. In order to keep the population of APCs at steady state, a replenishing mechanism is included. It is proportional to the difference between the current concentration APCi(t), and the generic steady state value 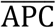 , divided by the absolute value of the same difference plus a modulating constant that we set = 1, i.e.,
, divided by the absolute value of the same difference plus a modulating constant that we set = 1, i.e., 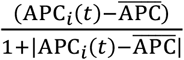 . The rationale behind the previous definition is that if the concentration APCi(t) is close to
. The rationale behind the previous definition is that if the concentration APCi(t) is close to 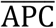 the term is essentially zero, whereas if the difference is large, the denominator is equally large, and the ratio is close to one. This allows a near constant modulation rate that only acts whenever the population of macrophages is far from equilibrium. This reequilibration rate is denoted by GAPC. The APC population is also impacted by the interaction between APCs and invading viral particles. This causes the population of APCs to become activated at a rate TAPC, which is proportional to the product of the concentration of these cells and the viral load.
the term is essentially zero, whereas if the difference is large, the denominator is equally large, and the ratio is close to one. This allows a near constant modulation rate that only acts whenever the population of macrophages is far from equilibrium. This reequilibration rate is denoted by GAPC. The APC population is also impacted by the interaction between APCs and invading viral particles. This causes the population of APCs to become activated at a rate TAPC, which is proportional to the product of the concentration of these cells and the viral load.
Equations for URT Naïve APCs:
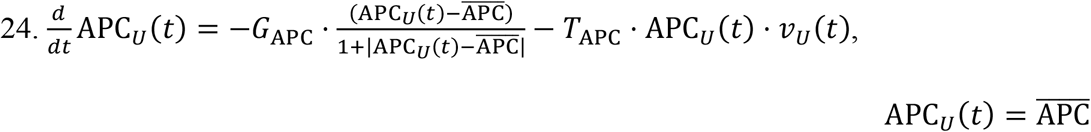 Equations for the LRT Naïve APCs:
Equations for the LRT Naïve APCs:
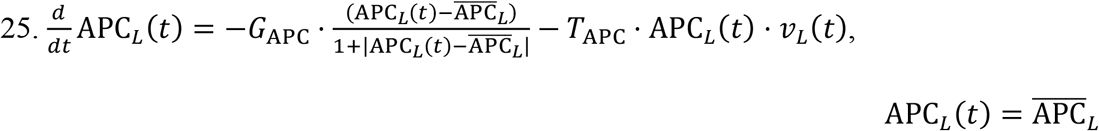
GI tract Naïve APCs:
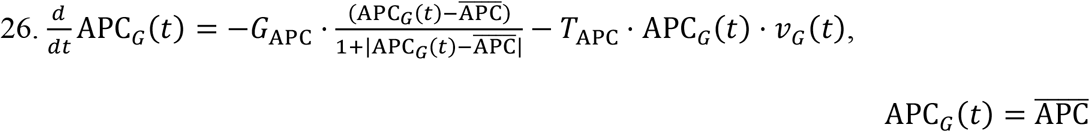
MPS Naïve APCs:
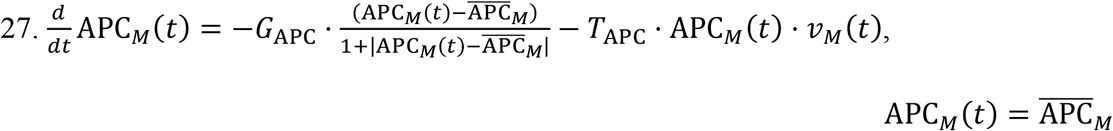
Heart Naïve APCs:
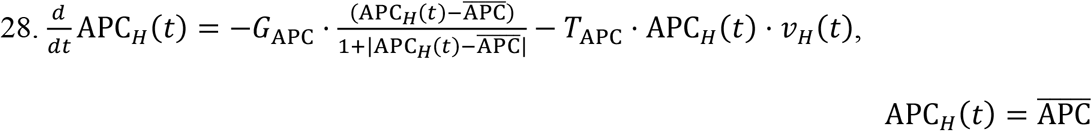
Kidney Naïve APCs:
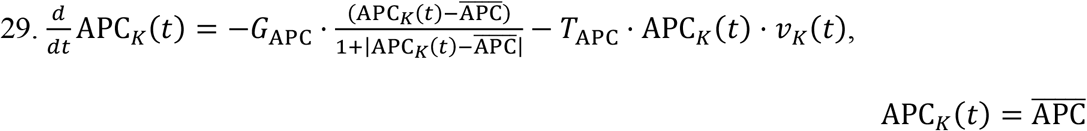
Brain Naïve APCs:
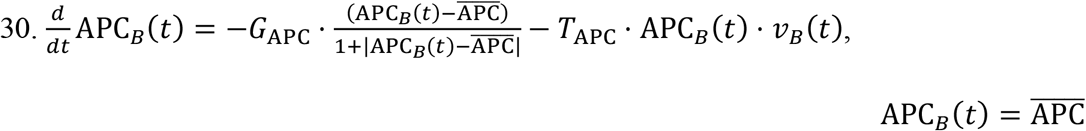
Lymphatic Activated APCs:
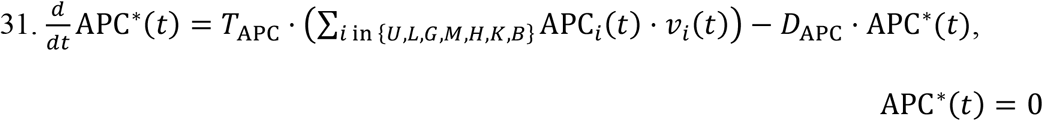
As mentioned earlier, the concentration of activated APCs, denoted by APC∗(t), increases at a rate proportional to the conversion rate TAPC of naïve APCs to the activated state. At the same time, activated APCs can become incapacitated or simply eliminated after a certain period of time. These processes are pooled into the APC death rate constant DAPC.
The remaining equations in the model describe either lymphocyte or antibody concentrations. The lymphocytes under consideration are CD8+, CD4+, and B cells. The concentration of the naïve or inactivated version of these populations is represented by CD8(t), CD4(t), and BC(t), respectively. Similar to the APC case, the steady state concentration for each of these cell types is denoted with a horizontal bar on top of each variable. This value is maintained using a mechanism analogous to the APC case, i.e., through the term 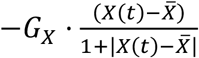 , where X is the corresponding cell type.
, where X is the corresponding cell type.
Furthermore, each of these lymphocytes can be promoted to its activated state, which we denote with an asterisk, X∗. This conversion takes place when activated APCs interact with naïve lymphocytes, presenting them fragments of the ingested viral particles. The transformation rate from the naïve state to the activated one is represented by the constant TX, where X indicates the cell type. For the case of CD4+ and CD8+, we use the common rate TT. The concentration of activated lymphocytes can fluctuate due to two mechanisms. One is the conversion of naïve cells, which was already discussed. The second is due to death caused by a variety of factors like exhaustion or apoptosis, and is expressed through the death constant DX. B cells, however, follow a different mechanism, which we discuss later.
Naïve CD8:
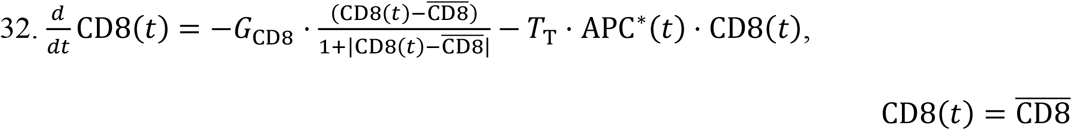
Active CD8*:
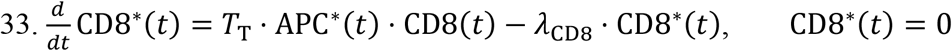
Naïve B cells:
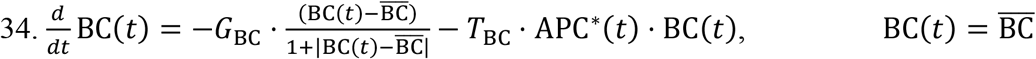
Activated B cells:
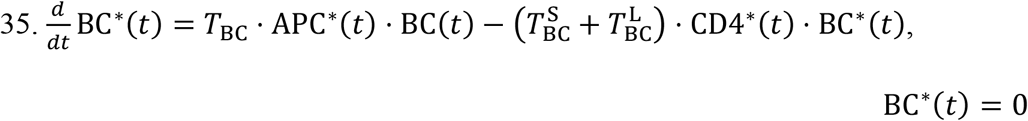
Activated B cells alone cannot neutralize the viral load. They must first transform into plasma cells, which then in turn produce antibodies that can carry out the viral neutralization. The transition from activated B cell to plasma cell is enabled by activated CD4* cells. This transformation can generate two types of plasma cell: short-lived (S) and long-lived (L). The transition rates are represented by the constants 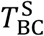 and
and 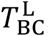 , and the corresponding concentrations of plasma cells is denoted by PS(t) and PL(t), respectively. Lastly, each type possesses a particular death rate, indicated by the constant
, and the corresponding concentrations of plasma cells is denoted by PS(t) and PL(t), respectively. Lastly, each type possesses a particular death rate, indicated by the constant 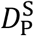 for the short-lived type and
for the short-lived type and 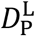 for the long-lived type.
for the long-lived type.
Naïve CD4:
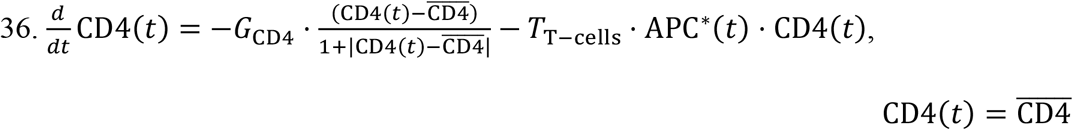
Activated CD4:
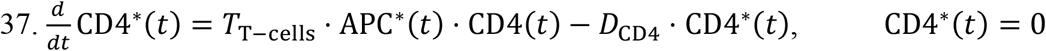
Short-lived plasma cells:
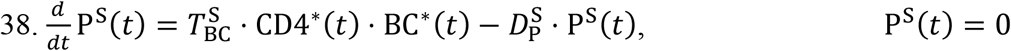
Long-lived plasma cells:
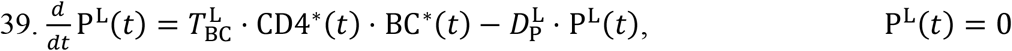
Antibodies:
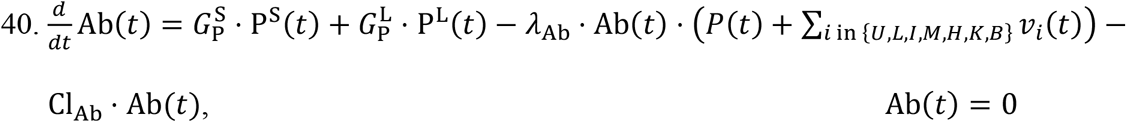
The final equation in the model describes the rate of change of antibody concentration, Ab(t). The sole contributors to the production of antibodies are short-lived and long-lived plasma cells. The corresponding production rates are given by 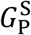 and
and 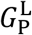 , respectively. We consider two mechanisms by which antibodies can be consumed or eliminated. One is the loss of antibodies due to their interaction with viral particles in a given compartment with rate constant λAb. The second is the elimination of antibodies by other factors independent of the viral load, e.g., degradation or clearance from tissues, which occurs at rate constant CLAb. A summary of the parameters used in the model is given in Tables 1-3.
, respectively. We consider two mechanisms by which antibodies can be consumed or eliminated. One is the loss of antibodies due to their interaction with viral particles in a given compartment with rate constant λAb. The second is the elimination of antibodies by other factors independent of the viral load, e.g., degradation or clearance from tissues, which occurs at rate constant CLAb. A summary of the parameters used in the model is given in Tables 1-3.
2. Parametric analysis
Once the full set of parameters for the model has been defined and fixed for the conditions corresponding to Figure 4, we proceed to identify the relative effect of each parameter on the viral kinetics. This is done by systematically perturbing one or multiple parameters and comparing the outcomes between the perturbed state and the original state. To quantify the differences between the two states we introduce the following 6 criteria. The first three refer to the area under the curve (AUC) of the viral load curves in the URT, the LRT, and the Plasma compartment for a period of 30 days. Mathematically,
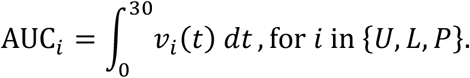
The next three denote the total time required for the viral load in the URT, LRT, and Plasma compartment to reduce to < 102 viral copies per mL, i.e., 2 in log base 10 units. The last comparison criterion is the antibody titer at day 30 in the body, i.e., Ab(t = 30). In the next two subsections we compare each of the 6 criteria using global sensitivity analysis (GSA), applied to the 31 parameters.
Global sensitivity analysis
Local sensitivity analysis only allows one parameter to change at a time, and thus may not be suitable for analyzing complex biological systems. We performed global sensitivity analysis (GSA36, 54, 55; i.e., multiple parameters can be varied simultaneously) to obtain a further understanding of the relationship between parameters and their combined effects on the model outputs of interest. Specifically, in our analysis, all parameters were perturbed at the same time, and we assume a uniform distribution for each parameter. The range of values of each sample was PR ± 99%(PR), where PR is the reference value of a given parameter. To ensure a complete sampling of the full parameter space we used a Latin hypercube sampling scheme. Subsequently, each sample was used to recompute the kinetics, from which we extract the 6 criteria mentioned above. In total, 10 batches of 5000 samples each were computed. Then, the data were subjected to a multivariate linear regression analysis to produce an impact factor, which we label as the sensitivity index (SI). Lastly, the sensitivity indices were ranked by means of a one-way ANOVA and a Tukey’s test. The higher the SI, the more significance a parameter holds. A detailed description of the GSA workflow can be found in38, 60, 61. All analyses were performed in MATLAB R2018a.
Data Availability
The manuscript uses and references data from published literature.
Author Contributions
PD, ZW conceived the study, conceptualized the idea, and designed the model. PD, JRR developed the model. PD, JRR, KS, MJP, MR collected the data. PD, KS, JDB, VKY, RP, WA, MB, DS, VC, ZW interpreted the data. PD, JRR, JDB, ZW performed the model analysis. PD, JRR, KS, JDB, MJP, MR, RP, WA, DS, VC, ZW wrote or edited the manuscript.
Competing Interests
The authors have declared that no competing interests exist.
Acknowledgements
This research has been supported in part by the National Science Foundation Grant DMS-1930583 (VC, ZW), the National Institutes of Health (NIH) Grants 1U01CA196403 (VC, ZW), 1U01CA213759 (VC, ZW), 1R01CA226537 (RP, WA, VC, ZW), 1R01CA222007 (VC, ZW), and U54CA210181 (VC, ZW). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Author affiliations updated
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.
- 11.
- 12.↵
- 13.↵
- 14.
- 15.
- 16.
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.
- 38.↵
- 39.
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.
- 63.
- 64.
- 65.
- 66.
- 67.
- 68.
- 69.
- 70.
- 71.
- 72.
- 73.
- 74.
- 75.
- 76.
- 77.
- 78.
- 79.
- 80.
- 81.
- 82.
- 83.
- 84.
- 85.
- 86.
- 87.
- 88.
- 89.



