
Abstract
Following its emergence in late 2019, SARS-CoV-2 has caused a global pandemic resulting in unprecedented efforts to reduce transmission and develop therapies and vaccines (WHO Emergency Committee, 2020; Zhu et al., 2020). Rapidly generated viral genome sequences have allowed the spread of the virus to be tracked via phylogenetic analysis (Hadfield et al., 2018; Pybus et al., 2020; Worobey et al., 2020). While the virus spread globally in early 2020 before borders closed, intercontinental travel has since been greatly reduced, allowing continent-specific variants to emerge. However, within Europe travel resumed in the summer of 2020, and the impact of this travel on the epidemic is not well understood. Here we report on a novel SARS-CoV-2 variant, 20A.EU1, that emerged in Spain in early summer, and subsequently spread to multiple locations in Europe, accounting for the majority of sequences by autumn. We find no evidence of increased transmissibility of this variant, but instead demonstrate how rising incidence in Spain, resumption of travel across Europe, and lack of effective screening and containment may explain the variant’s success. Despite travel restrictions and quarantine requirements, we estimate 20A.EU1 was introduced hundreds of times to countries across Europe by summertime travellers, likely undermining local efforts to keep SARS-CoV-2 cases low. Our results demonstrate how genomic surveillance is critical to understanding how travel can impact SARS-CoV-2 transmission, and thus for informing future containment strategies as travel resumes.
CAVEATS
20A.EU1 most probably rose in frequency in multiple countries due to travel and difference in SARS-CoV-2 prevalence. There is no evidence that it spreads faster.
There are currently no data to evaluate whether this variant affects the severity of the disease.
While dominant in some countries, 20A.EU1 has not taken over everywhere and diverse variants of SARS-CoV-2 continue to circulate across Europe. 20A.EU1 is not the cause of the European ‘second wave.’
SARS-CoV-2 is the first pandemic where the spread of a viral pathogen has been globally tracked in near real-time using phylogenetic analysis of viral genome sequences (Hadfield et al., 2018; Pybus et al., 2020; Worobey et al., 2020). SARS-CoV-2 genomes continue to be generated at a rate far greater than for any other pathogen and more than 200,000 full genomes are available on GISAID as of November 2020 (Shu and Mc-Cauley, 2017).
In addition to tracking the viral spread, these genome sequences have been used to monitor mutations which might change the transmission, pathogenesis, or antigenic properties of the virus. One mutation in particular, D614G in the spike protein, has received much attention. This variant (Nextstrain clade 20A) seeded large outbreaks in Europe in early 2020 and subsequently dominated the outbreaks in the Americas, thereby largely replacing previously circulating lineages. This rapid rise has led to the suggestion that this variant is more transmissible (Korber et al., 2020; Volz et al., 2020), which is corroborated by experimental studies (Plante et al., 2020; Yurkovetskiy et al., 2020).
Following the global dissemination of SARS-CoV-2 in early 2020 (Worobey et al., 2020), intercontinental travel dropped dramatically. Within Europe, however, travel and in particular holiday travel resumed in summer (though at lower levels than in previous years) with largely uncharacterized effects on the pandemic. Here we report on a novel SARS-CoV-2 variant 20A.EU1 (S:A222V) that emerged in early summer 2020, presumably in Spain, and subsequently spread to multiple locations in Europe. Over the summer, it rose in frequency in parallel in multiple countries. As we report here, this variant, 20A.EU1, and a second variant 20A.EU2 with mutation S477N in the spike protein account for the majority of recent sequences in Europe.
Recently emerged variants in Europe
Figure 1 shows a time scaled phylogeny of sequences sampled in Europe and their global context, highlighting the variants in this manuscript. Clade 20A and its daughter clades 20B and 20C have variant S:D614G and are colored in yellow. A cluster of sequences in clade 20A has an additional mutation S:A222V colored in orange. We designate this cluster as 20A.EU1 (it has since also been labeled as lineage B.1.177).
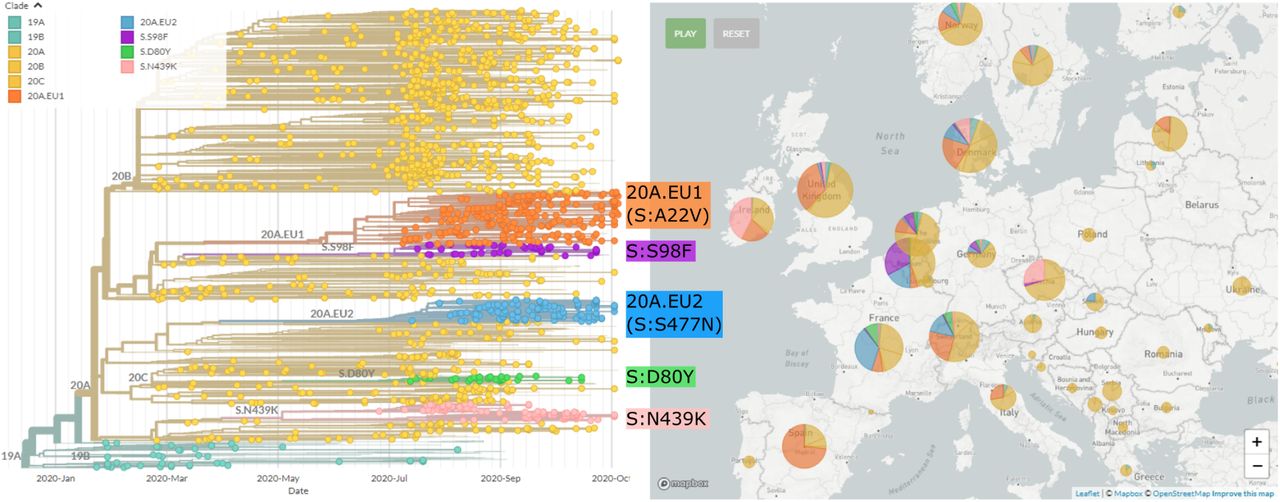
Phylogenetic overview of SARS-CoV-2 in Europe. The tree shows a representative sample of isolates from Europe colored by clade and by the variants highlighted in this paper. A novel variant (orange; 20A.EU1) with mutation S:A222V on a S:D614G background emerged in early summer and is common in most countries with recent sequences. A separate variant (20A.EU2, blue) with mutation S:S477N is prevalent in France. On the right, the proportion of sequences belonging to each variant (through the duration of the pandemic) is shown per country. Tree and visualization were generated using the Nextstrain platform (Hadfield et al., 2018) as described in methods.
In addition to the 20A.EU1 cluster we describe here, an additional variant (20A.EU2; blue in Fig. 1) with several amino acid substitutions, including S:S477N and mutations in the nucleocapsid protein, has become common in some European countries, particularly France (Fig. S5). The S:S477N substitution has arisen multiple times independently, for example in a variant in clade 20B that has dominated the recent outbreak in Oceania. The position 477 is close to the receptor binding site (Fig. S1), and deep mutational scanning studies indicate that S:S477N slightly increases the receptor binding domain’s affinity for ACE2 (Starr et al., 2020). Moreover, the SARS-CoV-2 spike residue S477 is part of the epitope recognized by the C102 neutralizing antibody (Barnes et al., 2020) and the detection of multiple variants at this position, such as S477N, might have resulted from the selective pressure exerted by the host immune response.
Several other smaller clusters defined by the spike mutations D80Y, S98F, N439K are also seen in multiple countries (see Table I and Fig. S5). While none of these have reached the prevalence of 20A.EU1 or 20A.EU2, some have attracted attention in their own right: S:N439K has appeared twice in the pandemic (Thomson et al., 2020), is found across Europe (particularly Ireland and the UK), is located in the RBD, and is an escape mutation from antibody C135 (Barnes et al., 2020; Weisblum et al., 2020) and S:Y453F, also in the RBD, has appeared multiple times, may be an adaptation to mink (Rodrigues et al., 2020), is also an escape mutation for an antibody (Baum et al., 2020), and was associated with an outbreak in Danish mink. Focal phylogenies for these, and other variants mentioned in this paper, can be found at nextstrain.org/groups/neherlab.
Representative mutations of 20A.EU1 (the focus of this study) and other notable variants.
Updated phylogenies of SARS-CoV-2 in Europe and individual European countries are provided at nextstrain.org/groups/neherlab. The page also includes links to analyses of the individual clusters discussed here.
Functional characterization of S:A222V
Our analysis here focuses on the variant 20A.EU1 with substitution S:A222V. S:A222V is in the spike protein’s domain A (Figure S1) also referred to as the NTD) (Mc-Callum et al., 2020; Tortorici et al., 2020), which is not known to play a direct role in receptor binding or membrane fusion for SARS-CoV-2. However, mutations can sometimes mediate long-range effects on protein conformation or stability.
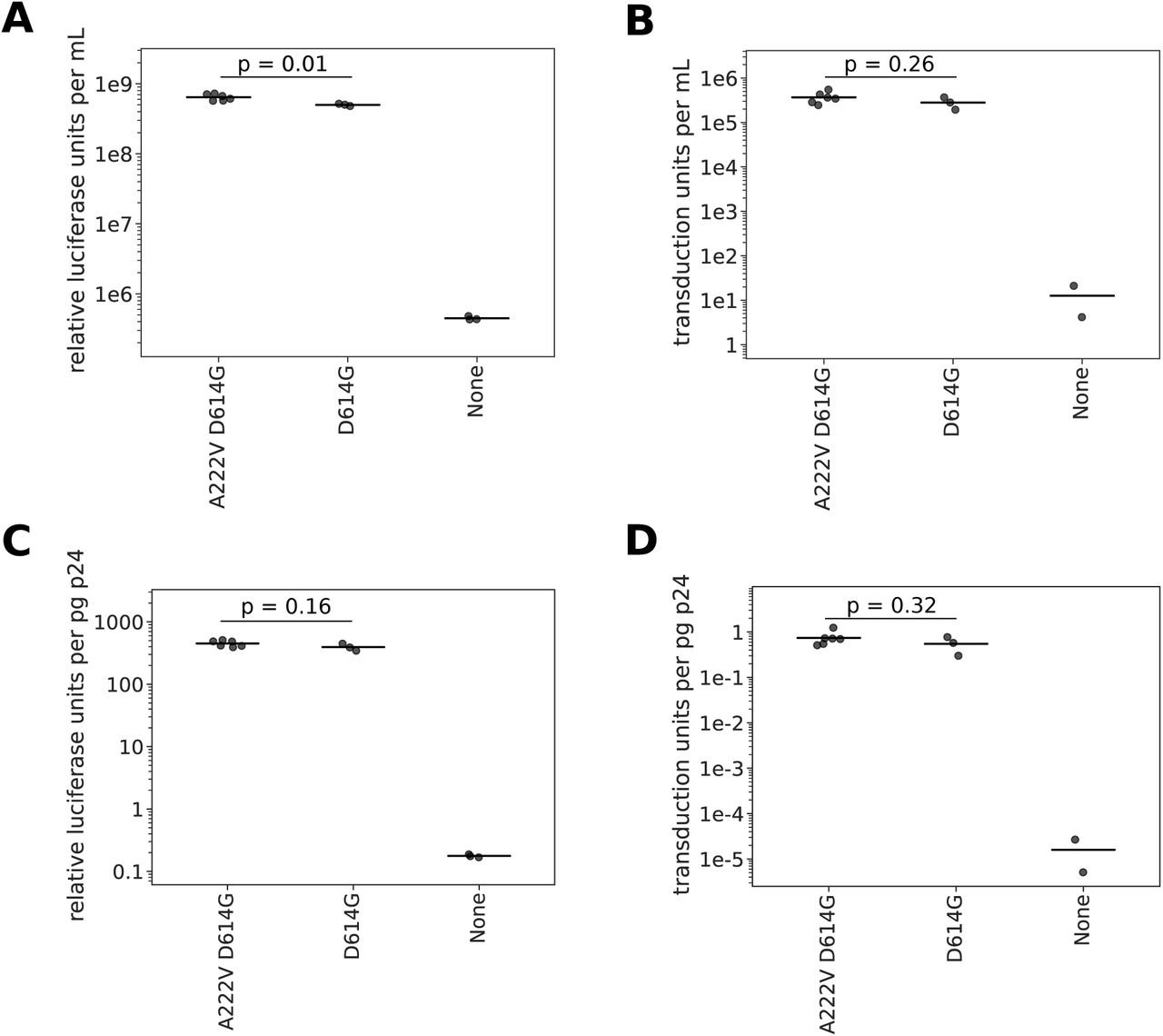
To test whether the S:A222V mutation had an obvious functional effect on spike’s ability to mediate viral entry, we produced lentiviral particles pseudotyped with spike either containing or lacking the A222V mutation in the background of the D614G mutation and deletion of the end of spike’s cytoplasmic tail. Lentiviral particles with the A222V mutant spike had slightly higher titers than those without (mean 1.3-fold higher), although the difference was not statistically significant (Fig. S2). Therefore, A222V does not lead to the same large increases in the titers of spike-pseudotyped lentiviral that has been observed for the D614G mutation (Korber et al., 2020; Yurkovetskiy et al., 2020), which is a mutation that is now generally considered to have increased the fitness of SARS-CoV-2 (Plante et al., 2020; Volz et al., 2020). However, we note that this small effect must be interpreted in equivocal terms, as the effects of mutations on actual viral transmission in humans are not always paralleled by measurements made in highly simplified experimental systems such as the one used here. Therefore, we examined epidemiological and evolutionary evidence to assess if the variant showed evidence of enhanced transmissibilty in humans.
Early observations of 20A.EU1
The earliest sequences identified date from the 20th of June, when 7 Spanish sequences and 1 Dutch sequence were sampled. The next non-Spanish sequence was from the UK (England) on the 18th July, with a Swiss sequence sampled on the 22nd and an Irish sampled on the 23rd. By the end of July, samples from Spain, the UK (England, Northern Ireland), Switzerland, Ireland, Belgium, and Norway were identified as being part of the cluster. By the 22nd August, the cluster also included sequences from France, Denmark, more of the UK (Scotland, Wales), Germany, Latvia, Sweden, and Italy. Two sequences from Hong Kong, three from Australia, fifteen from New Zealand, and six sequences from Singapore, presumably exports from Europe, were first detected in mid-August (Hong Kong, Australia), mid-September (New Zealand), and mid-October (Singapore).
The proportion of sequences from several countries which fall into the cluster, by ISO week, is plotted in Fig. 2, showing how the cluster-associated sequences have risen in frequency (Fig 2). The cluster first rises in frequency in Spain, initially jumping to around 60% prevalence within a month of the first sequence being detected. In the United Kingdom, France, Ireland, and Switzerland we observe a gradual rise starting in mid-July. In Wales and Scotland the variant was at 80% by mid-September (Fig S3), whereas frequencies in Switzerland and England were around 50% at that time. In contrast, Norway observed a sharp peak in early August, but few sequences are available for later dates. The date ranges and number of sequences observed in this cluster are summarized in Table SI.

We include the eight countries which have at least 20 sequences from 20A.EU1. The symbol size indicates the number of available sequence by country and time point in a non-linear manner. Travel restrictions are shown to/from Spain, as this is the possible origin of the cluster. Most European countries allowed quarantine-free travel to other (non-Spanish) countries in Europe for a longer period. When the last data point included only very few sequences, it has been dropped for clarity.
Cluster Source and Number of Introductions across Europe
Fig. 3 shows a collapsed phylogeny, as described in Methods, indicating the observations of different geno-types within the 20A.EU1 cluster across Europe. The prevalence of early samples in Spain, diversity of the Spanish samples, and prominence of the cluster in Spanish sequences suggest Spain as the likely origin for the cluster, or at least the place where it first expanded and became common. Epidemiological data from Spain indicates the earliest sequences in the cluster are associated with two known outbreaks in the north-east of the country. The cluster variant seems to have initially spread among agricultural workers in Aragon and Catalonia, then moved into the local population, where it was able to travel to the Valencia Region and on to the rest of the country (though sequence availability varies between regions). This initial expansion may have been critical in increasing the cluster’s prevalence in Spain just before borders re-opened.
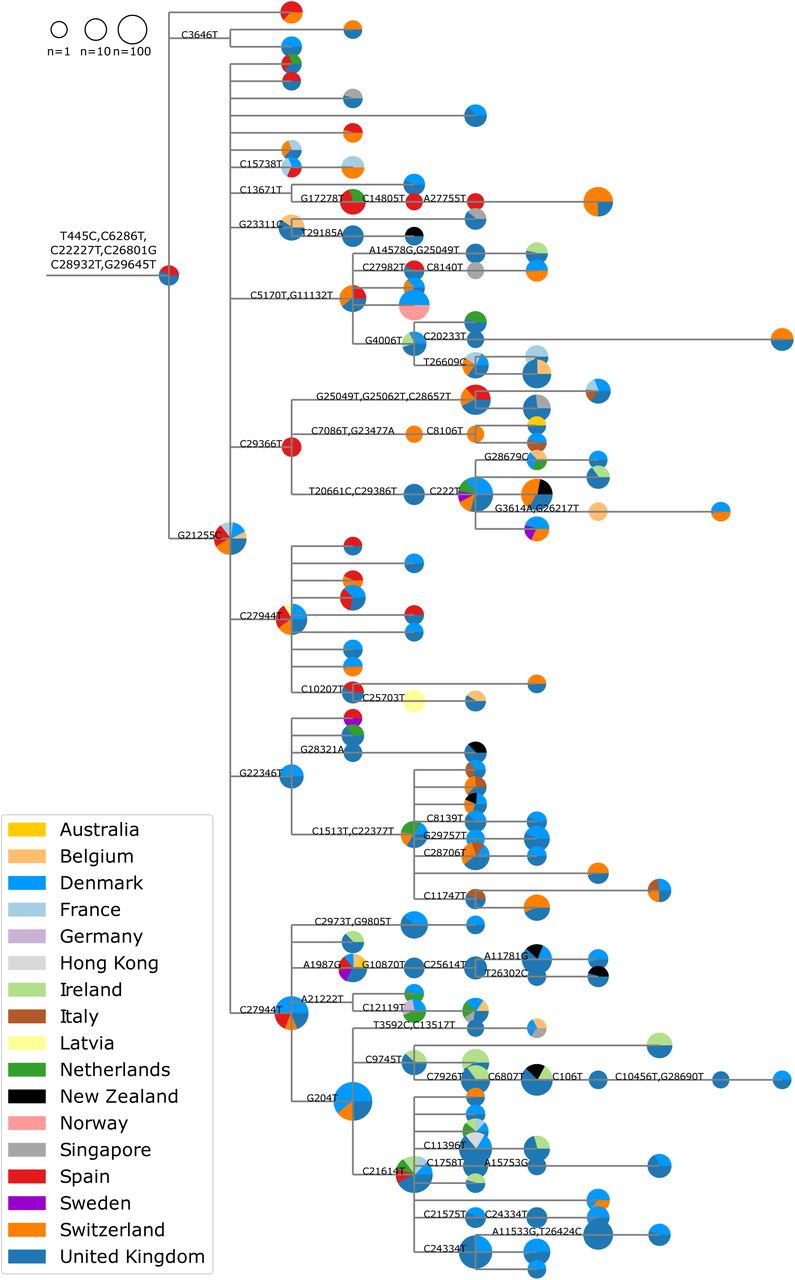
The phylogeny shown is the subtree of the 20A.EU1 cluster, with sequences carrying all six defining mutations. Pie charts show the representation of sequences from each country at each node. Size of the pie chart indicates the total number of sequences at each node. Pie chart fractions scale non-linearly with the true counts (fourth root) to ensure all countries are visible.
Since it is unlikely that diversity and phylogenetic patterns sampled in multiple countries arose independently, it is reasonable to assume that the majority of mutations within the cluster arose once and were carried (possibly multiple times) between countries. We use this rationale to provide lower bounds on the number of introductions to different countries. Throughout July and August 2020, Spain had a higher per capita incidence than most other European countries (see Fig S7) and 20A.EU1 was much more prevalent in Spain then elsewhere, suggesting Spain as likely origin of most 20A.EU1 importations. We therefore assume that genotypes sampled in Spain arose in Spain. However, the 256 sequences in the cluster from Spain likely do not represent the full diversity. Variants found only outside of Spain may reflect diversity that arose in secondary countries, or may represent diversity not sampled in Spain. In particular, as the UK sequences much more than any other country in Europe, it is not unlikely they may have sampled diversity that exists in Spain but has not yet been sampled there. Despite limitations in sampling, Fig. 3 clearly shows that most major genotypes in this cluster were distributed to multiple countries, suggesting that many countries have experienced multiple introductions of identical genotypes that cannot be resolved. Finally, while initial introductions of the variant likely originated from Spain, phylogenetic analysis suggests that later transmissions involved other European countries (see Fig. 3 and 20A.EU1 Nextstrain build online).
Per-Country Inferences
In some cases only a single 20A.EU1 genotype was sampled in a country, but in many countries multiple distinct genotypes were sampled, indicating multiple introductions, and these we will cover in more detail below. There are 26 non-European samples in the cluster, from Hong Kong, Australia, New Zealand, and Singapore. All are likely exports from Europe: the Hong Kong sequences indicate a single introduction, whereas the Australian, Singaporean, and New Zealand samples are from at least two, six, and seven separate transmissions, respectively, from Europe. Interestingly, seven of the sequences from New Zealand appear to be linked to in-flight transmission en-route to New Zealand, likely originating from two passengers from Switzerland (Swadi et al., 2020).
Many EU and Schengen-area countries, including Switzerland, the Netherlands, and France, opened their borders to other countries in the bloc on 15th June, though the Netherlands kept the United Kingdom on their ‘orange’ list. Spain opened its borders to EU member states (except Portugal, at Portugal’s request) and associated countries on 21st June.
Norway, Latvia, Germany, Italy, Sweden
The sequences from Norway, Latvia, and Germany all indicate single introduction events, whereas Sweden and Italy’s sequences indicate at least four and six introductions, respectively. Germany, Sweden, and Italy have only a small number of sequences – two, seven, and ten, respectively – meaning that many introductions might have been missed. Norway and Latvia’s larger sequence counts form two clear separate monophyletic groups within the 20A.EU1 cluster. The Norwegian samples seem likely to be a direct introduction from Spain, as they cluster tightly with Spanish sequences and the first sample (29th July) was just after quarantine-free travel to Spain was stopped. In Latvia, quarantine-free travel to Spain was only allowed until the 17th July - a month before the first sequence was detected on 22nd August. Latvia allowed quarantine-free travel to other European countries for a longer period, and this introduction may therefore have come via a third country.
Switzerland
Quarantine-free travel to Spain was possible from 15th June to 10th August. The majority of holiday return travel is expected from mid-July to mid-August towards the end of school holidays. When all lineages circulating in Switzerland since 1 May are considered, the notable rise and expansion of 20A.EU1 is clear (see Fig S6).
To estimate introductions, we consider 19 genotypes observed in Switzerland that are also observed in Spain or directly descend from a genotype observed in Spain, suggesting an introduction into Switzerland, directly from Spain or indirectly, through a third country. Additionally, we see 14 nodes where a genotype was observed in Switzerland and in another non-Spanish country, suggesting either an additional import from Spain, a third country, or a transmission between Switzerland and the other country. Three of the 33 nodes involve more than twenty Swiss sequences, and seem to have grown rapidly, consistent with the growth of the overall cluster.
For those nodes that don’t directly or through their parents share diversity with Spanish sequences, the Swiss sequences are most closely related to diversity found in the UK, France, and Denmark, suggesting possible transmission between other EU countries and Switzerland or diversity in Spain that was not sampled.
Belgium
Along with many European countries, Belgium reopened to EU and Schengen Area countries on the 15th June. Belgium employed a regional approach to travel restrictions, meaning that while travellers returning from some regions of Spain were subject to quarantine from the 6th of August, it was not until the 4th September that most of Spain was subject to travel restrictions. Belgian sequences share diversity with sequences from Spain, the UK, Denmark, and the Netherlands, and France, among others, spread across 9 nodes in the phylogeny. Three of these nodes share diversity with Spanish sequences, or descend from nodes with Spanish sequences.
France
France has had no restrictions on EU and Schengen-area travel since it re-opened borders on the 15th of June. France’s 32 sequences cluster across nine nodes on the phylogeny: in three nodes the sequences cluster with Spanish sequences and four nodes stem directly from a parent with Spanish sequences. The remaining two nodes are genetically further from the diversity sampled in Spain, and may indicate an introduction from another country, possibly the United Kingdom or Switzerland.
Netherlands
The Netherlands began imposing a quarantine on travellers returning from some regions of Spain on the 28th July, increasing the areas from which travellers must quarantine until the whole of Spain was included on the 25th August. Twelve nodes across the phylogeny contain sequences from the Netherlands. On three nodes sequences from the Netherlands share diversity with Spanish sequences, suggesting possible direct importations from Spain, and one node descends from a parent containing both Spanish and Dutch sequences. The earliest sample from the Netherlands was identified on the 20th June, the same date as the first sequences from Spain. However, travel began increasing from Spain to the Netherlands markedly earlier than to most European countries (Fig. 4 A), and this Dutch sequence nests within the diversity of early sequences from Spain, suggesting this sequence is the result of the earliest export of the variant outside of Spain.

Travel from Spain to other European countries resumed in July (though low compared to previous years). Assuming that travel returnees are infected at the average Spanish incidence of 20A.EU1 and transmit the virus at the rate of their resident population, imports from Spain are expected to account between 2 and 10% of SARS-CoV-2 cases after the summer.
Denmark
Denmark re-opened their borders to the majority of European countries on the 27th of June. By the end of July, however, the government was advising travellers to Spain’s Aragon, Catalonia, and Navarra regions to be tested for SARS-CoV-2 on their return. On the 6th of August, Denmark advised against all non-essential travel to Spain, and strongly suggested quarantine on return, though notably quarantine has not been a legal requirement, as it has been in many other countries in Europe. The 1,736 sequences from Denmark are found on 58 nodes across the phylogeny, with seven of these nodes containing both Danish and Spanish sequences, and 18 descending directly from nodes with Spanish sequences, suggesting multiple introductions of the 20A.EU1 variant.
The UK and Ireland
The first sequences in the UK (England) which associate with the cluster are from the 18th July, in the middle of the period from the 10th to 26th July when quarantine-free travel to Spain was allowed for England, Wales, and Northern Ireland. The first Irish sequences to associate with the cluster were taken a short time later, on the 23rd of July.
The large number of sequences from the United Kingdom make introductions harder to quantify. A total of 103 nodes in the phylogeny contain sequences from the United Kingdom. 15 of these nodes share diversity with Spanish sequences, while a further 28 descend directly from nodes that contain Spanish sequences. The remaining nodes most often share diversity with Denmark, Switzerland, and Ireland. Many of the nodes containing UK sequences are represented by dozens to hundreds of genomes, while one genotype present in the UK, carrying the 21614T mutation, is responsible for almost a half of the sequences associated with the cluster in the country.
The 83 sequences of the 20A.EU1 variant from Ireland cluster in 14 nodes on the phylogeny. In six nodes, Irish sequences either share diversity with Spanish sequences or have parents that do. Notably, every node containing Irish sequences also shares diversity with sequences from the United Kingdom. However, as mentioned before, the diversity in Spain is likely not fully represented in the tree, so direct transmission cannot be ruled out.
Differing Travel Restrictions in the UK and Ireland
While quarantine-free travel was allowed in England, Wales, and Northern Ireland from the 10th–26th July, Scotland refrained from adding Spain to the list of ‘exception’ countries until the 23th July (meaning there were only 4 days during which returnees did not have to quarantine). On the other hand, Ireland never allowed quarantine-free travel to Spain, but did allow quarantine-free travel from Northern Ireland. Similarly, Scotland allowed quarantine-free travel to and from England, Wales, and Northern Ireland. Despite having only a very short or no period where quarantine-free travel was possible from Spain, both Scotland and Ireland have cases linked to the cluster consistent with significant travel volume between Spain and these countries over the summer. Additionally, close connections to the UK countries with similarly high travel volumes may have allowed further introductions.
No evidence for transmission advantage of 20A.EU1
During a dynamic outbreak, it is particularly difficult to unambiguously tell whether a particular variant is increasing in frequency because it has an intrinsic advantage, or because of epidemiological factors (Grubaugh et al., 2020). In fact, it is a tautology that every novel big cluster must have grown recently and multiple lines of independent evidence are required in support of an intrinsically elevated transmission potential.
The cluster we describe here – 20A.EU1 (S:A222V) – was dispersed across Europe initially mainly by travelers to and from Spain. To explore whether repeated imports are sufficient to explain the rapid rise in frequency and the displacement of other variants, we estimated the expected contribution of imports given the passenger volume and the incidence in Spain and other European countries (see Fig. 4). The number of confirmed cases in Spain rose from around 10 cases per 100k inhabitants per week in early July to 100 in late August. Taking reported incidence at face value and assuming that returning tourists have a similar incidence, we expect more than 800 introductions into the UK (see Table SII and Fig. 4 for tourism summaries (Instituto Nacional de Estadistica, 2020) and departure statistics (Aena.es, 2020)). Similarly, Switzerland would expect around 160 introductions. A simple model that tracks these imports and their subsequent local spread over the summer in the resident epidemics in different countries in Europe predicts that the frequencies of 20A.EU1 would start rising in July, continue to rise through August, and be stable thereafter in concordance with observations in many countries including Switzerland, Denmark, France, Wales, and Scotland (see Fig. 4 B).
While the shape of the expected frequency trajectories from imports in Fig. 4 B is consistent with observations, this naive import model underestimates the final frequency of 20A.EU1 by a factor between 2 and 13. Given the simplicity of the model, no quantitative match should be expected.
The overall impact of imported variants depends on several uncertain factors such as the relative ascertainment rate in source and destination populations, the probability that travelers are exposed, and the propensity of travel returnees to transmit further. SARS-CoV-2 incidence in holiday destinations, and in the locations where travelers return, may not be well-represented by the national averages used in the model. For example, during the first wave in spring, some ski resorts had exceptionally high incidence and contributed disproportionately to dispersal of SARS-CoV-2. Furthermore, the risk of exposure and onward transmission are likely increased by travel-related activities both abroad and at home. Travel precautions such as quarantine should in principle prevent spread of SARS-CoV-2 infections acquired abroad, but in practice compliance may have been imperfect.
To investigate the possibility of faster growth of 20A.EU1 introductions, we identified 20A.EU1 and non-20A.EU1 introductions into Switzerland and their down-stream Swiss transmission chains (see Methods). Over-all, we identify 14-84 introductions of the 20A.EU1 variant. Phylodynamic estimates of the effective reproductive number (Re) through time for introductions of 20A.EU1 and for other variants (see Fig. S8) suggest a tendency for 20A.EU1 introductions to (transiently) grow faster. This signal of faster growth, however, is more readily explained with increased travel-associated transmission than intrinsic differences to the virus. Indeed, the frequency of 20A.EU1 plateaued in most countries after the summer travel period, consistent with import driven dynamics with little or no competitive advantage. Only in England did its frequency continue to increase after the main summer travel period ended (Fig. S3), though for many countries recent data are lacking.
Comparatively high incidence over the summer of non-20A.EU1 variants and hence a relatively low impact of imported variants (e.g. Belgium, see Fig. S7) might explain why 20A.EU1 remains at low frequencies in some countries despite high-volume travel to Spain. To date, 20A.EU1 has not been observed in Russia, consistent with little travel to/from Spain and continuously high SARS-CoV-2 incidence.
Notably, case numbers across Europe started to rise rapidly around the same time the 20A.EU1 variant started to become prevalent in multiple countries, (Fig. S4). However, countries where 20A.EU1 is rare (Belgium, France, Czech Republic - see Fig. S5) have seen similarly rapid increases, suggesting that this rise was not driven by any particular lineage and that 20A.EU1 has no difference in transmissibility. Furthermore, we observe in Switzerland that Re increased in fall by a comparable amount for the 20A.EU1and non-20A.EU1variants (see (Fig. S8). The arrival of fall and seasonal factors are a more plausible explanation for the resurgence of cases (Neher et al., 2020).
DISCUSSION
The rapid spread of 20A.EU1 and other variants underscores the importance of a coordinated and systematic sequencing effort to detect, track, and analyze emerging SARS-CoV-2 variants. In many countries we do not know which variants are circulating now since little recent sequence data are available, and it is only through multi-country genomic surveillance that it has been possible to detect and track this and other variants.
The rapid rise of these variants in Europe highlights the importance of genomic surveillance of the SARS-CoV-2 pandemic. If any mutations are found to increase the transmissibility of the virus, previously effective infection control measures might no longer be sufficient. Along similar lines, it is imperative to understand whether novel variants impact the severity of the disease. So far, we have no evidence for any such effect: the low mortality over the summer in Europe was pre-dominantly explained by a much better ascertainment rate and a marked shift in the age distribution of confirmed cases. This variant was not yet prevalent enough in July and August to have had a big effect. As sequences and clinical outcomes for patients infected with this variant become available, it will be possible to better infer whether this lineage has any impact on disease prognosis.
Finally, our analysis highlights that countries should carefully consider their approach to travel when large-scale inter-country movement resumes across Europe. Whether the 20A.EU1 variant described here has rapidly spread due to a transmission advantage or due to epidemiological factors alone, its observed repeated introduction and rise in prevalence in multiple countries implies that the summer travel guidelines and restrictions were generally not sufficient to prevent onward transmission of introductions. While long-term travel restrictions and border closures are not tenable or desirable, identifying better ways to reduce the risk of introducing variants, and ensuring that those which are introduced do not go on to spread widely, will help countries maintain often hard-won low levels of SARS-CoV-2 transmission.
Data Availability
All sequence data is available on GISAID (gisaid.org), and the code for the analyses performed is available on Github and linked in the manuscript text.
Transparency declaration
DV is a consultant for Vir Biotechnology Inc. The Veesler laboratory has received an unrelated sponsored research agreement from Vir Biotechnology Inc. The other authors declare no competing interests.
Authors’ contribution
EBH identified the cluster, led the analysis, and drafted the manuscript. RAN analysed data and drafted the manuscript. MZ and SN analysed data and created figures. VD investigated structural aspects and created figures. JDB and KC performed lentiviral assays and created figures. IC and FGC interpreted the origin of the cluster and contributed data. All authors contributed to and approved the final manuscript.
METHODS
Phylogenetic analysis
We use the Nextstrain pipeline for our phylogenetic analyses https://github.com/nextstrain/ncov/ (Hadfield et al., 2018). Briefly, we align sequences using mafft (Katoh et al., 2002), subsample sequences (see below), add sequences from the rest of the world for phylo-genetic context based on genomic proximity, reconstruct a phylogeny using IQ-Tree (Minh et al., 2019) and infer a time scaled phylogeny using TreeTime (Sagulenko et al., 2018). For computational feasibility, ease of interpretation, and to balance disparate sampling efforts between countries, the Nextstrain-maintained runs sub-sample the available genomes across time and geography, resulting in final builds of ∼4,000 genomes each.
Sequences were downloaded from GISAID using the nextstrain/ncov workflow on the 10th November 2020. A table acknowledging the invaluable contributions by many labs is available as a supplement. The Swiss SARS-CoV-2 sequencing efforts are described in (Nadeau et al., 2020) and (Stange et al., 2020). The majority of Swiss sequences used here are from the Nadeau et al. (2020) data set, the remainder are available on GISAID.
Defining the 20A.EU1 Cluster
The cluster was initially identified as a monophyletic group of sequences stemming from the larger 20A clade with amino acid substitutions at positions S:A222V, ORF10:V30L, and N:A220V or ORF14:L67F (overlapping reading frame with N), corresponding to nucleotide mutations C22227T, C28932T, and G29645T. In addition, sequences in 20A.EU1 differ from their ancestors by the synonymous mutations T445C, C6286T, and C26801G. There are currently 19,695 sequences in the cluster by this definition.
The sub-sampling of the standard Nextstrain analysis means that we are not able to visualise the true size or phylogenetic structure of the cluster in question. To specifically analyze this cluster using almost all available sequences, we designed a specialized build which focuses on cluster-associated sequences and their most genetically similar neighbours. For computational reasons, we limit the number of samples to 900 per country per month. As only the UK has more sequences than this per month, this results in a random down-sampling of sequences from the UK for the months of August, September, and October. Further, we excluded several problematic sequences: France/BR8951/2020 for very high intra-sample variation, England/PORT-2D2111/2020 and England/LIVE-1DD7AC/2020 for one confirmed and one suspected wrong date (divergence values do not match the given date), and 92 Irish sequences with inaccurate dates (confirmed with the submitter).
We identify sequences in the cluster based on the presence of nucleotide substitutions at positions 22227, 28932, and 29645 and use this set as a ‘focal’ sample in the nextstrain/ncov pipeline. This selection will exclude any sequences with no coverage or reversions at these positions, but the similarity-based sampling during the Nextstrain run will identify these, as well as any other nearby sequences, and incorporate them into the dataset. We used these three mutations as they included the largest number of sequences that are distinct to the cluster. By this criterion, there are currently 19,436 sequences in the cluster – slightly fewer than above because of missing data at these positions.
To visualise the changing prevalence of the cluster over time, we plotted the proportion of sequences identified by the four substitutions described above as a fraction of the total number of sequences submitted, per ISO week. Frequencies of other clusters are identified in an analogous way.
Phylogeny and Geographic Distribution
The size of the cluster and number of unique mutations among individual sequences means that interpreting overall patterns and connections between countries is not straightforward. We aimed to create a simplified version of the tree that focuses on connections between countries and de-emphasizes onward transmissions within a country. As our focal build contains ‘background’ sequences that do not fall within the cluster, we used only the monophyletic clade containing the four amino-acid changes and three synonymous nucleotide changes that identify the cluster. Then, subtrees that only contain sequences from one country were collapsed into the parent node. The resulting phylogeny contains only mixed-country nodes and single-country nodes that have mixed-country nodes as children. Nodes in this tree thus represent ancestral genotypes of subtrees: sequences represented within a node may have further diversified within their country, but share a set of common mutations. We count all sequences in the subtrees towards the geographic distribution represented in the pie-charts in Fig. 3.
This tree allows us to infer lower bounds for the number of introductions to each country, and to identify plausible origins of those introductions. It is important to remember that, particularly for countries other than the United Kingdom, the full circulating diversity of the variant is probably not being captured, thus intermediate transmissions cannot be ruled out. In particular, the closest relative of a particular sequence will often have been sampled in the UK simply because sequencing efforts in the UK exceed most other countries by orders of magnitude. It is, however, not our goal to identify all introductions but to investigate large scale patterns of spread in Europe.
Estimation of contributions from imports
To estimate how the frequency of 20A.EU1 is expected to change in country X due to travel, we consider the following simple model: A fraction αi of the population of X returns from Spain every week i (estimated from travel data (Aena.es, 2020)) and is infected with 20A.EU1 with a probability pi given by its per capita 7 day incidence in Spain. The week-over-week fold change of the epidemic within X is calculated as gi = (ci − αipi)/ci−1, where ci is the per capita incidence in week i in X. The total number of 20A.EU1 cases vi in week i is hence vi = givi−1 +αi, while the total number of non-20A.EU1 cases is ri = giri−1. Running this recursion from mid-June to November results in the frequency trajectories in Fig. 4.
Phylodynamic analysis of Swiss transmission chains
We identified introductions into Switzerland and down-stream Swiss transmission chains by considering a tree of all available Swiss sequences combined with foreign sequences with high similarity to Swiss sequences (full procedure described in Nadeau et al. (2020)). Putative transmission chains were defined as majority Swiss clades allowing for at most 3 “exports” to third countries. Identification of transmission chains is complicated by polytomies in SARS-CoV-2 phylogenies and we bounded the resulting uncertainty by either (i) considering all substrees descending from the polytomy as separate introductions and (ii) aggregating all into a single introduction, see (Nadeau et al., 2020) for details.
The phylodynamic analysis of the transmission chains was performed using BEAST2 with a birth-death-model tree prior (Bouckaert et al., 2019; Stadler et al., 2013). 20A.EU1 and non-20A.EU1 variants share a sampling probability and log Re has an Ornstein-Uhlenbeck prior, see Nadeau et al. (2020) for details.
Pseudotyped Lentivirus Production and Titering
The S:A222V mutation was introduced into the protein-expression plasmid HDM-Spike-d21-D614G, which encodes a codon-optimized spike from Wuhan-Hu-1 (Genbank NC 045512) with a 21-amino acid cytoplasmic tail deletion and the D614G mutation (Greaney et al., 2020). This plasmid is also available on AddGene (plasmid 158762). We made two different versions of the A222V mutant that differed only in which codon was used to introduce the valine mutation (either GTT or GTC). The sequences of these plasmids (HDM Spike-d21-D614G-A222V-GTT and HDM Spike-d21-D614G-A222V-GTC) are available as supplement files at github.com/emmahodcroft/cluster scripts/.
Spike-pseudotyped lentiviruses were produced as described in (Crawford et al., 2020). Two separate plasmid preps of the A222V (GTT) spike and one plasmid prep of the A222V (GTC) spike were each used in duplicate to produce six replicates of A222V spike-pseudotyped lentiviruses. Three plasmid preps of the initial D614G spike plasmid (with the 21-amino acid cytoplasmic tail truncation) were each used once used to make three replicates of D614G spike-pseudotyped lentiviruses. All viruses were titered in duplicate.
Lentiviruses were produced with both Luciferase IRES ZsGreen and ZsGreen only lentiviral backbones (Crawford et al., 2020), and then titered using luciferase signal or percentage of fluorescent cells, respectively. All viruses were titered in 293T-ACE2 cells (BEI NR-52511) as described in (Crawford et al., 2020), with the following modifications. Viruses containing luciferase were titered starting at a 1:10 dilution followed by 5 serial 2-fold dilutions. The Promega BrightGlo luciferase system was used to measure relative luciferase units (RLUs) ∼ 65 hours post-infection and RLUs per mL were calculated at each dilution then averaged across all dilutions for each virus. Viruses containing only ZsGreen were titered starting at a 1:3 dilution followed by 4 serial 5-fold dilutions. The 1:375 dilution was visually determined to be ∼ 1% positive about 65 hours post-infection and was used to calculate the percent of infected cells using flow cytometry (BD FACSCelesta cell analyzer). Viral titers were then calculated using the percentage of green cells via the Poisson formula. To normalize viral titers by lentiviral particle production, p24 concentration (in pg/mL) was quantified by ELISA according to kit instructions (Advanced Bioscience Laboratories Cat. #5421). All viral supernatants were measured in technical duplicate at a 1:100,000 dilution.
Data availability
All code used for the above analyses is available at github.com/emmahodcroft/cluster_scripts (the commit tagged journal submission was used to generate the figures in this manuscript). The code used to run the cluster builds is available at github.com/emmahodcroft/ncov cluster. Sequence data were obtained from GISAID and tables listing all accession numbers of sequences are available as supplementary information.
SUPPLEMENTARY MATERIAL
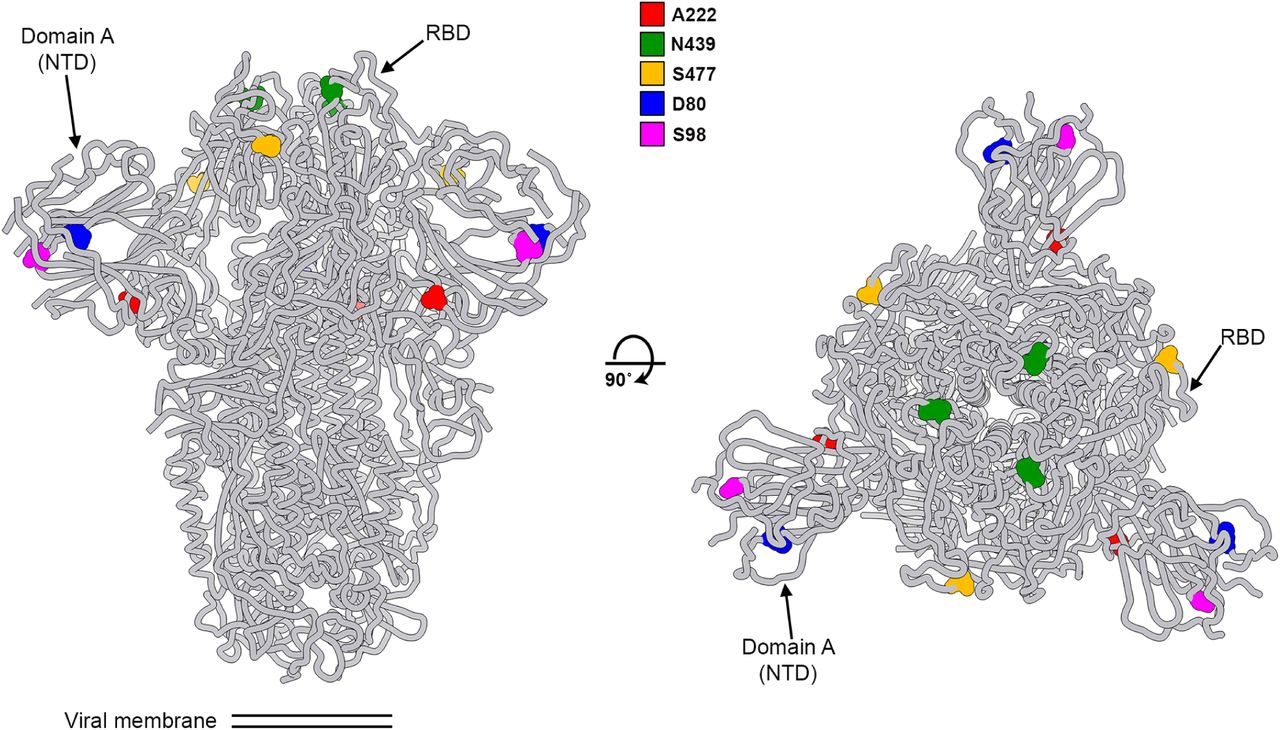
Two orthogonal orientations of the SARS-CoV-2 spike glycoprotein trimer highlighting the position of the variants described in the manuscript. 222: red; 439: green; 477: orange; 80: blue; 98: magenta
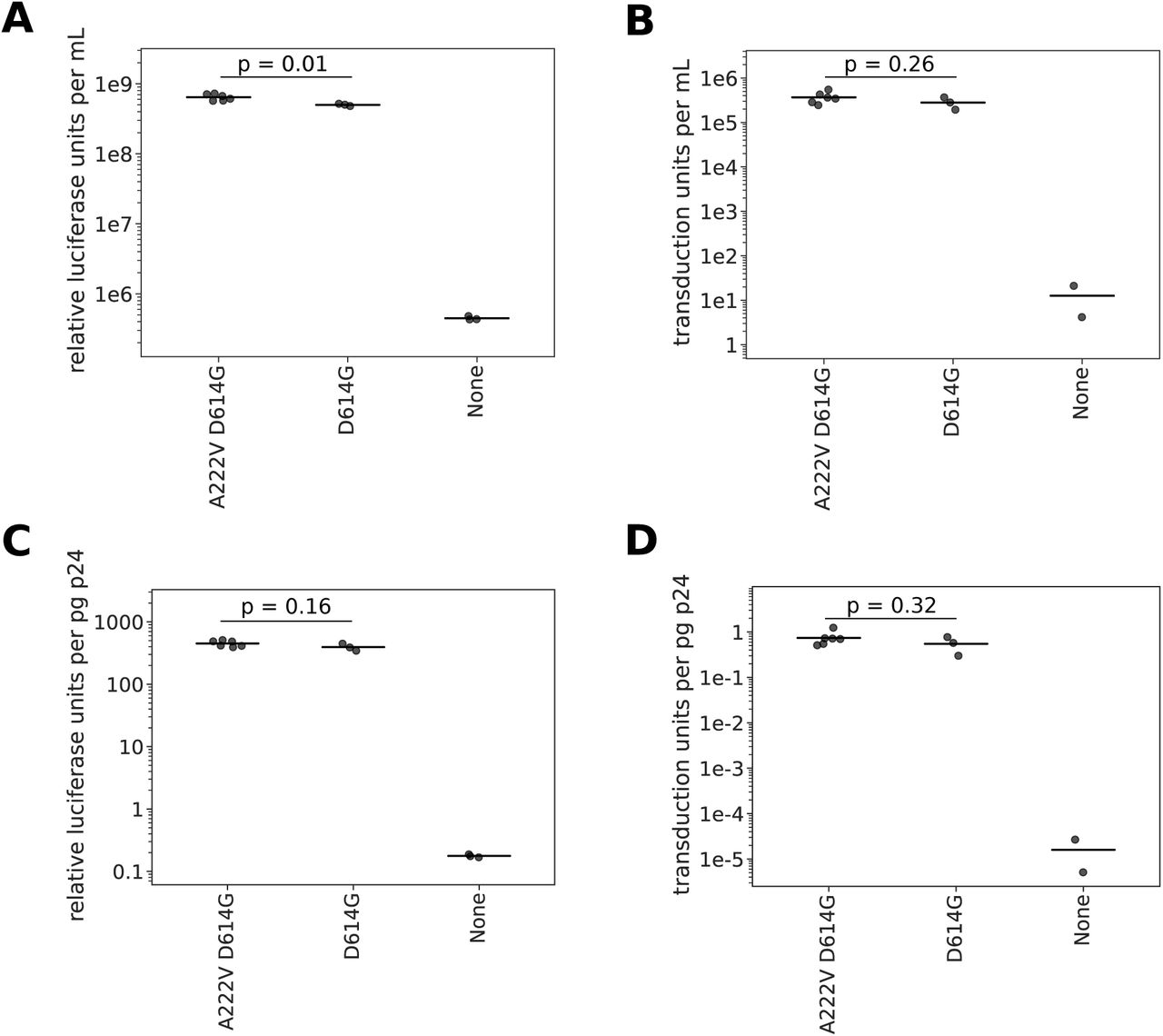
Titers of lentiviral particles pseudotyped with spike with or without the A222V mutation. A) Titers of lentiviral particles carrying luciferase in the viral genome. The horizontal line indicates the mean titer. B) Titers of lentiviral particles carrying the fluorescent protein ZsGreen in the viral genome. The horizontal line indicates the mean titer. C) Titers of lentiviral particles carrying luciferase in the viral genome normalized by the p24 concentration (pg/mL) of each viral supernatant. D) Titers of lentiviral particles carrying ZsGreen in the viral genome normalized by the p24 concentration (pg/mL) of each viral supernatant. All p-values calculated using a two-sided t-test.
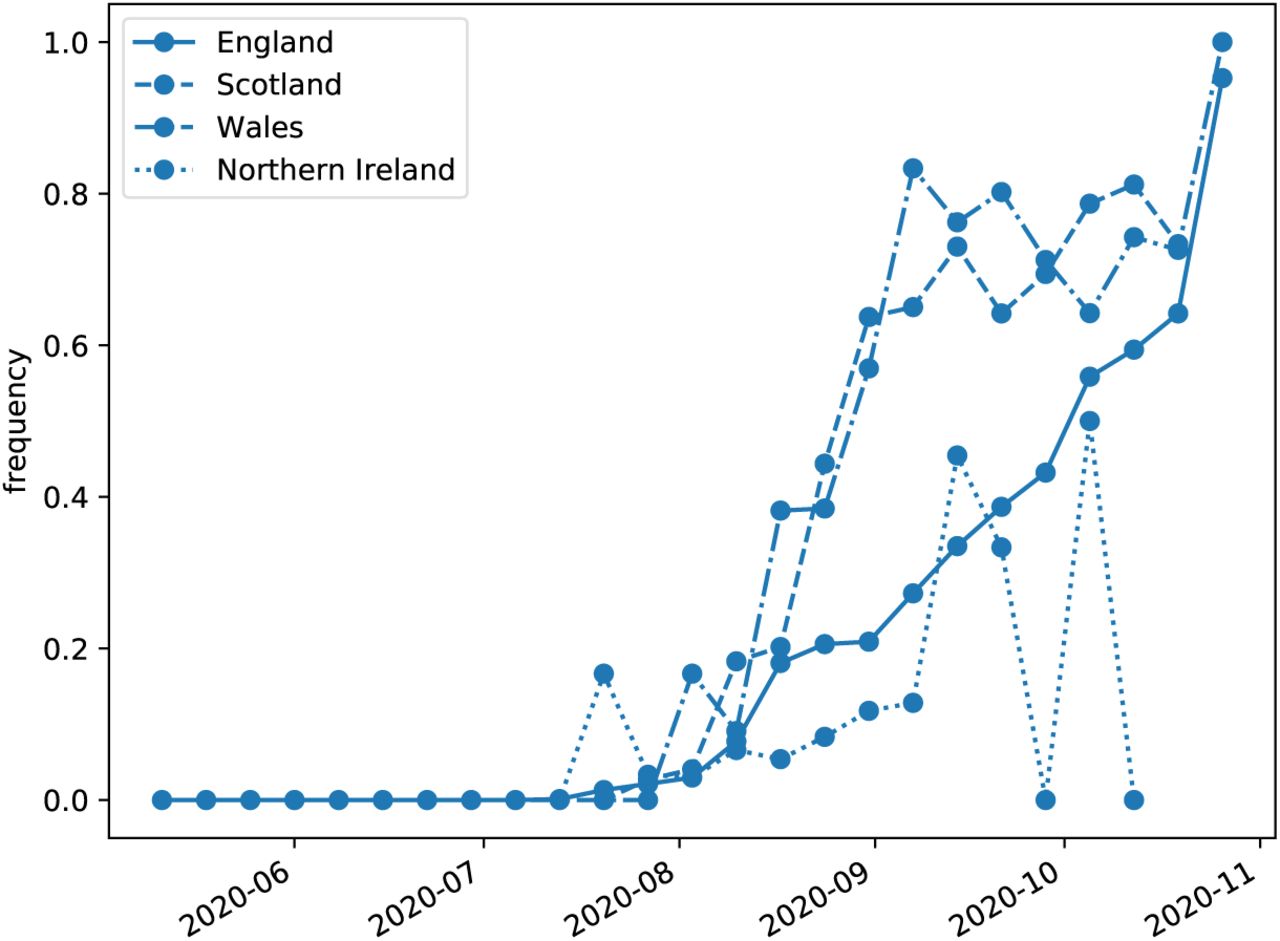
Frequency of submitted samples from the constituent countries of the United Kingdom that fall within the cluster.

Sequence availability varies by country, but Spain, Switzerland, Denmark, and the United Kingdom have provided data until October-November. In the United Kingdom, Denmark, and Switzerland, the novel variant was first detected in July-August and rose quickly through August and September. Case data were obtained from ECDC (European Center for Disease Control, 2020).

In countries with at least ten sequences that fall into any of the defined clusters, the proportion of sequences per ISO week that fall into each cluster is shown.
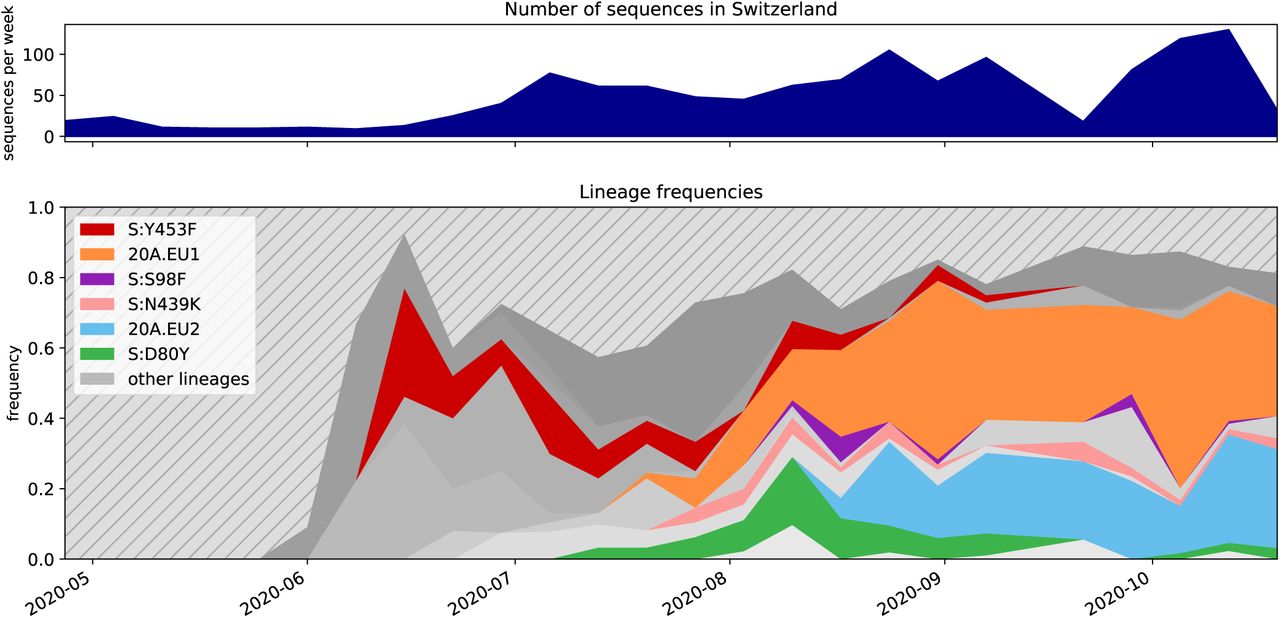
Lineages found in a Swiss-focused Nextstrain build. A lineage is defined as a node present in the tree after the cutoff date of 1 May 2020 with at least 10 Swiss sequences as children. Clusters discussed in this manuscript are labelled. Lineages are shown as the proportion of the total number of sequences per week in Switzerland. Striped space in the bottom graph represents lineages with most recent common ancestors dating back prior to 1 May 2020 and lineages that do not contain at least 10 Swiss sequences.

Incidence in various countries over the summer. Spain and Belgium had relatively higher incidence from the start of July compared with other countries in Europe.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
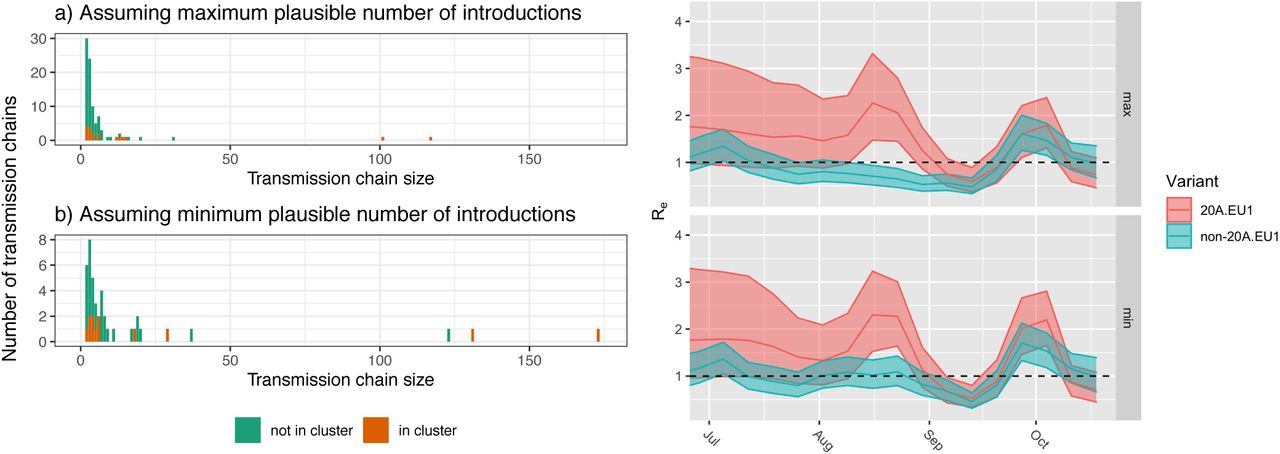
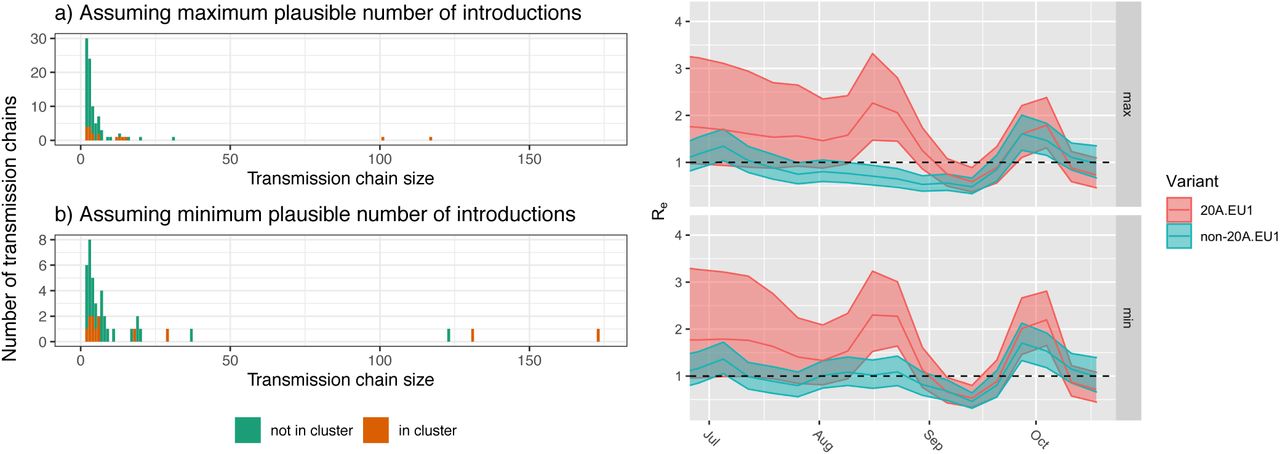
Introduction and spread of the 20A.EU1-variant in Switzerland. (a,b) The size of transmission chains caused by introductions into Switzerland under two extreme definitions of an introduction. Not shown are the number of singletons, which are introductions with no evidence of onward transmission. Depending on the min/max definition of introductions, there were between 2 or 66 singletons of 20A.EU1 (14 or 78% of all 20A.EU1 introductions) and 15 or 264 non 20A.EU1 (33 or 79% of all non-20A.EU1 introductions). 20A.EU1 introductions are slightly bigger on average, but the difference is not significant. (c,d) The effective reproductive number estimated for 20A.EU1 (red) and the non-20A.EU1 variants (green) under the two extreme definitions of an introduction. While there is little data to inform estimates of Re for 20A.EU1 in July and it differs little from the prior, there is some evidence that 20A.EU1 was growing faster than other variants in August. However, systematic differences in ascertainment in travel associated cases might confound this inference. Starting September, Re of 20A.EU1 is statistically indistinguishable from that of other variants. Shaded areas indicate 95% HPD regions.
Summary of sequences observed in the 20A.EU1 cluster, sorted by the date of the first observed sequence.
Arrival statistics of tourists in Spain over the Summer 2020 (Instituto Nacional de Estadistica, 2020)
Acknowledgements
We are gratefully to researchers, clinicians, and public health authorities for making SARS-CoV-2 sequence data available in a timely manner. We also wish to thank the COVID-19 Genomics UK consortium for their notable sequencing efforts, which have provided more than half of the sequences currently publicly available. This work was supported by the SNF through grant numbers 31CA30 196046 (to RAN, EBH), 31CA30 196267 (to TS), core funding by the University of Basel and ETH Zürich, the National Institute of General Medical Sciences (R01GM120553 to DV), the National Institute of Allergy and Infectious Diseases (DP1AI158186 and HHSN272201700059C to DV), a Pew Biomedical Scholars Award (DV), an Investigators in the Pathogenesis of Infectious Disease Awards from the Burroughs Wellcome Fund (DV and JDB), a Fast Grants (DV), and NIAID grants R01AI141707 (JDB) and F30AI149928 (KHDC). SeqCOVID-SPAIN is funded by the Instituto de Salud Carlos III project COV20/00140, Spanish National Research Council and ERC StG 638553 to IC and BFU2017-89594R from MICIN to FGC. JDB is an Investigator of the Howard Hughes Medical Institute.
Footnotes
↵12 on behalf or the SeqCOVID-SPAIN consortium
↵14 SeqCOVID-SPAIN consortium
This update includes all new sequence data available until 10 Nov 2020, and analyses, descriptions, and figures have been updated accordingly. Additionally, we include details of lentiviral lab work investigating the S:A222V mutation, and a simple model illustrating how travel volume and SARS-CoV-2 prevalence can largely explain the spread of 20A.EU1 observed across Europe over the summer. None of the updates have changed the overall conclusions of the paper.
REFERENCES



