
Abstract
Coronavirus Disease 2019 (COVID-19) is a global pandemic caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). While molecular assays are used to detect viral genetic material for the diagnosis of acute infection, reliable serological assays are needed to measure immunity against SARS-CoV-2. In this report, we describe an enzyme-linked immunosorbent assay (ELISA) that detects antibodies against the following SARS-CoV-2 recombinant proteins: the full-length spike (S) protein and the receptor-binding domain (RBD). Our assay is sensitive and specific for immunoglobulin (Ig) G, IgA and IgM anti-S protein and anti-RBD antibodies. Samples were pre-treated with Triton X-100 to inactivate potential virus without affecting antibody detection. Our in-house ELISA performed as well as the commercial EUROIMMUN and Ortho assays for anti-SARS-CoV-2 antibodies. This method provides a high-throughput assay that does not require specialized instrumentation and can be widely used to determine immunity and the dynamic range of antibodies found within SARS-CoV-2.
Introduction
Coronavirus disease 2019 (COVID-19) is caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [1]. Containment of the virus has been challenged by asymptomatic and pre-symptomatic spread of SARS-CoV-2 [2]. Approximately 20% of symptomatic infections are severe and disproportionately impact the elderly population and patients with underlying health conditions [3, 4]. Symptoms of SARS-CoV-2 infection typically appear 2-14 days after viral exposure; however, asymptomatic carriers can transmit the virus [5]. It is estimated that up to 80% of people with COVID-19 have mild or asymptomatic disease and asymptomatic transmissions account for half of all COVID-19 cases [6-8]. Thus, serology is essential to capture actual infection rates as it captures both asymptomatic and symptomatic infections.
The laboratory diagnosis of COVID-19 is made using real-time reverse-transcription polymerase chain reaction (RT-PCR) detection of viral mRNA using nasal or throat swabs [9-12]. In the 4 days before the typical time of symptom onset, the probability of a false-negative result by RT-PCR decreases from 100% to 67% [13]. Viral RNA is detectable as early as the first day of symptoms and peaks within the first week of symptom onset [10]. RT-PCR positivity starts to decline by week 3 and subsequently becomes undetectable; however, PCR positivity may persist beyond three weeks in severely ill and hospitalized patients when most cases would yield a negative result [14]. Positive results reflect only the detection of viral RNA and do not necessarily indicate presence of viable virus, leading the assay to have a modest sensitivity (approximately 79%) and high specificity (approximately 100%) [9, 15, 16]. False-negative results can also be attributed to improper sample collection and timing of test [15, 17]. Accurate testing for SARS-CoV-2, followed by appropriate preventive measures, is paramount in the health care setting to prevent both nosocomial and community transmission. Currently, there is a need for tests that are capable of detecting the presence of SARS-CoV-2 specific antibodies with high accuracy to determine seroprevalence [18]. Although serology cannot distinguish between acute and chronic infection, it can identify individuals who have developed an immune response, aid in contact tracing, help identify suitable convalescent plasma donors, and can be used to evaluate vaccines.
The presence of antibodies to SARS-CoV-2 indicates the potential of protective immunity [19]. The target antigen of the antibody response to SARS-CoV-2 is a large glycoprotein termed spike (S) protein, which consists of the S1 and S2 domains. The S1 domain contains the receptor-binding domain (RBD), which is the target of most neutralizing antibodies [20-23]. The SARS-CoV-2 S protein is a homotrimer essential for mediating binding to host cells through interactions with the human receptor angiotensin converting enzyme 2 (ACE2) [20-22]. Binding of the RBD to the host ACE2 receptor triggers a conformational change in the S protein that subsequently initiates membrane fusion events with the host cell [24]. Although antibodies may be generated against multiple domains within the S protein, most neutralizing antibodies bind to the RBD [23, 25].
Antibody kinetics studies have reported that virus-specific IgG levels are lower in people who are asymptomatic or had a mild infection compared with people who had more severe symptoms and acute illness [26]. In this report, we describe a high-throughput ELISA assay with a high sensitivity and specificity for anti-SARS-CoV2 antibodies.
Methods and Materials
Patient Samples
Serum samples were collected from convalescent patients who had been confirmed to be symptomatic COVID-19 infection by RT-PCR (n=10, COVID-19-positive), COVID-19-negative individuals who tested negative by RT-PCR during the pandemic (n=4) and controls collected before November 2019 (pre-COVID controls, n=332). This study was approved by the Hamilton Integrated Research Ethics Board (HIREB) and informed written consent was obtained from all participants.
Production of S protein and RBD
A detailed protocol outlining protein production can be found in a study by Stadlbauer et al [27]. Plasmids encoding mammalian cell codon optimized sequences for SARS-CoV-2 full-length S protein and the RBD were generously gifted from the lab of Dr. Florian Krammer (Ichan School of Medicine at Mount Sinai, NY, NY, USA). In brief, proteins were produced in Expi293 cells (ThermoFisher, Waltham, MA, USA) using the manufacturer’s instructions. Post-transfection, when culture viability dropped to 40%, supernatants were collected and centrifuged at 500 x g for 5 minutes to remove cell debris. The supernatant was then incubated with shaking overnight at 4?C with 1 mL of nickel-nitrilotriacetic acid (Ni-NTA) agarose resin (Qiagen, Hilden, Germany) per 25 mL of transfected cell supernatant. The following day 10 mL polypropylene gravity flow columns (Qiagen) were used to elute the protein. RBD and S proteins were concentrated in Amicon centrifugal units (Millipore, Burlington, MA, USA), 10 kDa and 50 kDa respectively, prior to being resuspended in phosphate buffered saline (PBS). The purified proteins were analyzed via SDS-PAGE.
SARS-CoV-2 ELISA development
384 well plates (Nunc Maxisorp, Rochester, NY, USA) were coated overnight at 4°C with 25 μL/well of RBD (2 μg/mL) or S protein (5 μg/mL) suspended in 50 mM carbonate-bicarbonate buffer (pH 9.6). The plates were then blocked with 100 µL/well of 3% skim milk prepared in PBS with 0.05% Tween 20 at room temperature for 2 hours. The blocking solution was removed, and diluted patient serum samples (1/100 prepared in 1% skim milk in PBS/0.05% Tween 20) was added to the plates for 1 hour at room temperature. The plates were washed twice with PBS/0.05% Tween 20 and thrice with PBS. Bound human antibodies (IgG, IgA, or IgM) were detected with alkaline phosphatase conjugated goat anti-human IgG (γ-chain-specific, 1/2000, Jackson ImmunoResearch Laboratories, Inc, Westgrove, PA, USA), goat anti-human IgA (α-chain-specific; 1/500, Jackson ImmunoResearch Laboratories, Inc, Westgrove, PA, USA) antibody, or goat anti-human IgM (μ-chain-specific; 1/1000, Jackson ImmunoResearch Laboratories, Inc, Westgrove, PA, USA) antibody prepared in PBS/0.05% Tween 20. Plates were washed as before and followed with the addition of 50 µL substrate (4-nitrophenylphosphate disodium salt hexahydrate in diethanolamine; MilliporeSigma, St. Louis, MO, USA). The optical density at 405 nm and 490 nm (as a reference) was measured using a BioTek 800TS microplate reader (BioTek, Winooski, VT, USA). The cut-off was determined as the mean plus 2 standard deviations (SD) of the pre-COVID-19 control population. The specificities for each antigen/antibody combination were determined using the pre-COVID controls (n=332).
Measuring inter-assay variability and repeatability
To determine inter-assay reproducibility, pre-COVID-19 controls (n=4) and COVID-19 positive (n=4) patient serum samples were tested in at least four separate assays using the SARS-CoV-2 ELISA described above. Results were reported as an optical density (405 nm with reference 490 nm) for each antigen and antibody isotype (IgG, IgA and IgM). Values are represented as a ratio of observed optical density to the determined assay cut-off optical density. Values above 1 ratio is considered positive in the SARS-CoV-2 ELISA.
Evaluation of viral inactivation treatments on antibody testing in ELISA
Patient serum was inactivated to eliminate residual virus in serum samples. A subset of recovered COVID-19 positive (n=10) and pre-COVID-19 controls (n=5) were used to compare two viral inactivation methods in human sera: heat-treatment and treatment with the detergent Triton X-100 [28-30]. For heat-treatment, patient sera (0.5 mL) were incubated with rotational shaking at 56°C for 30 minutes and centrifuged for 10 minutes at 14,000 x g, after which the supernatant was collected [29]. In parallel, duplicate samples were mixed with Triton X-100 to a final concentration of 1% (v/v) [30]. The non-treated and treated patient samples were tested in the SARS-CoV-2 ELISA as previously described. We however did not test whether any virus was actually present in serum samples and whether the inactivation method was effective against virus.
Inhibition of IgG anti-RBD binding
To confirm the specificity of antibodies detected by the ELISA, we inhibited the binding of selected serum samples with excess fluid-phase RBD. One antibody-positive pre-COVID-19 control and COVID-19 positive (n=10) patient serum samples were tested in the SARS-CoV-2 ELISA described above. Samples diluted to a working concentration (1/100) were incubated with 10 times molar excess of RBD or the equivalent volume of PBS for 1 hour at room temperature before testing for antibodies in the standard assay as described.
Comparing the in-house SARS-CoV-2 ELISA to commercially available assays
A subset of COVID-19 positive patient samples (n=9) and COVID-19 negative patient samples (n=5) was tested in the commercially available EUROIMMUN Anti-SARS-CoV-2 ELISA that measures anti-S1 IgG and IgA and the results were compared to our in-house assay. The same set of COVID-19 positive and pre-COVID-19 controls was also tested by the Hamilton Regional Laboratory Medicine Program (HRLMP) in the Ortho Clinical Diagnostics COVID-19 IgG Antibody Test that measures anti-S protein IgG for comparison.
Statistical Analyses
Descriptive statistics were used to summarize the IgG, IgA, and IgM binding to S protein and RBD as measured by mean optical density across antigen replicates. Binomial 95% confidence intervals (CI) were calculated for all specificity determination. All statistical analyses were conducted using GraphPad Prism (version 7.0a, GraphPad Software, San Diego, USA).
Results
Patient Demographics
The pre-COVID-19 control population was made up of healthy individuals (n=26) and patients suspected of immune thrombocytopenia (ITP) or heparin induced thrombocytopenia (HIT) (n=306) with ages ranging between 21 and 79 years at time of collection. COVID-19 positive patient samples were collected between 23- and 89-days post-symptoms. COVID-19 negative patient samples were collected between 24- and 162-days post-symptoms.
ELISA development
We used a set of pre-COVID-19 controls and recovered COVID-19 positive patient samples to establish an ELISA with full-length S protein and RBD. The pre-COVID-19 controls (n=332) were used to establish the background reactivity to the S protein and RBD for IgG, IgA, and IgM antibodies, separately. The COVID-19 positive patients (n=14) were used to determine the reactivity of the ELISA to the full-length S protein and RBD. Antigen concentration of S protein and RBD and serum dilutions were optimized by testing COVID-19-positive and pre-COVD19 samples. We found that RBD at 2 μg/mL (Figure 1A) and S protein at 5 μg/mL (Figure 1B) were the optimal concentrations since these concentrations yielded the greatest separation between OD values of COVID-19 positive and pre-COVID-19 controls results (Figure 1). Saturation occurred at higher concentrations of each antigen. Using the same set of pre-COVID-19 controls and COVID-19 positive serum samples, we determined that 1/100 was the optimal serum dilution in 1% skim milk as it provided the best separation between negative and positive results (data not shown).

Binding of (A) anti-RBD and (B) anti-S protein IgG, A, and M was measured in the SARS-CoV-2 ELISA with plates coated at 1, 2, 5 or 10 μg/mL of the corresponding antigen. Black lines indicate recovered COVID-19 positive patient samples tested (n=8) and red lines represent pre-COVID-19 controls tested (n=8). The optimized concentrations selected were 2 μg/mL of RBD and 5 μg/mL of S-protein.
All recovered COVID-19 positive serum samples reacted strongly to both full-length S protein and RBD while reactivity of the pre-COVID-19 controls yielded lower levels of reactivity in most cases (below mean OD + 2SD cut-off). Reactivity of COVID-19 positive serum samples was stronger against the full-length S protein than against RBD.
Determining a cut-off for the SARS-CoV-2 ELISA
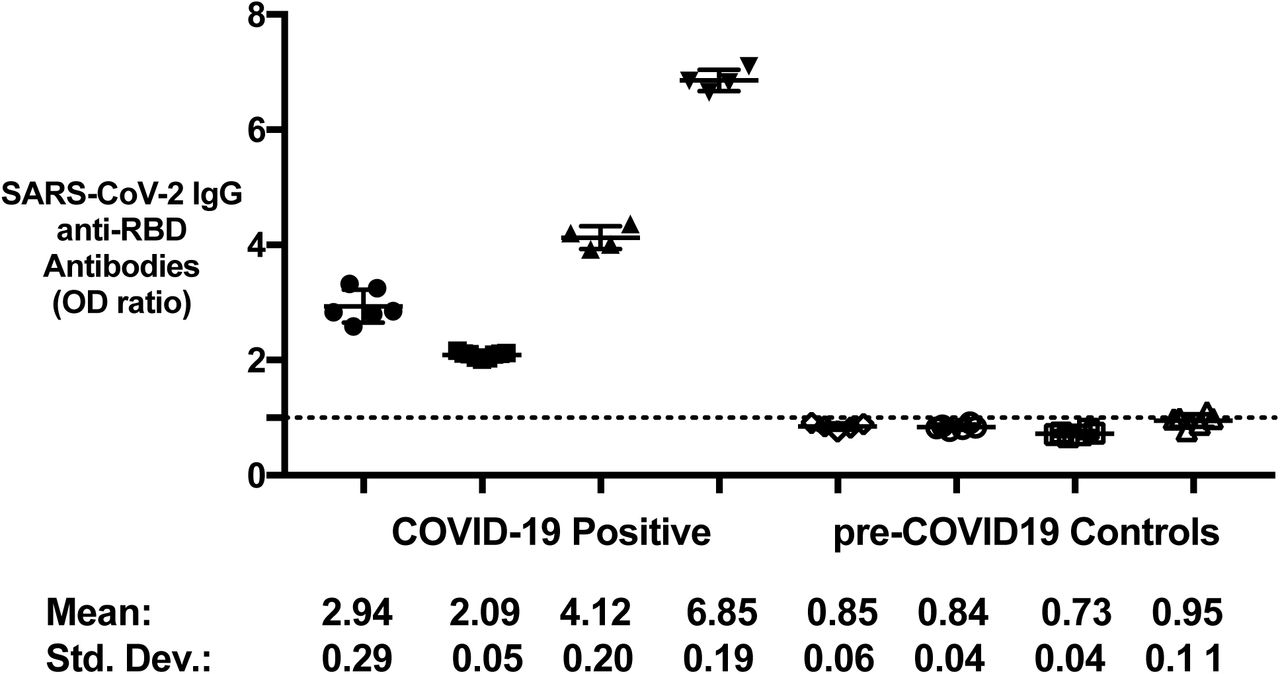
We used pre-COVID-19 controls (n=332) to determine the background reactivity and establish assay specificity to the S protein and RBD using samples from individuals drawn before the beginning of the pandemic. The cut-off was determined as the mean + 2SD of the OD readings in the pre-COVID-19 control population. We tested these samples using the optimized SARS-CoV-2 protocol. We found that most pre-COVID-19 controls had only background reactivity for both the full-length S protein and RBD (specificity for IgG = 96.7% and 97.0%, respectively). Each antigen and antibody isotype had a few pre-COVID-19 controls that tested positive for the antibodies based on the determined cut-off. We found that specificity was the lowest for IgM against both S protein and RBD, 94.6% (18 testing positive) and 95.5% (15 testing positive) respectively. S protein- and RBD-specific IgA had the highest measured specificity, 97.3% (9 testing positive) and 97.6% (8 testing positive), respectively (Figure 2).

A cut-off to determine what samples would be considered positive in the SARS-CoV-2 ELISA was determined by testing the binding of anti-RBD and anti-S protein IgG, A, and M of pre-COVID-19 controls (n=332). The cut-off was determined as the mean and 2 standard deviations for each isotype and antigen. Values are shown as a ratio of observed optical density to the determined assay cut-off optical density. Values above 1 ratio are considered positive in the SARS-CoV-2 ELISA.
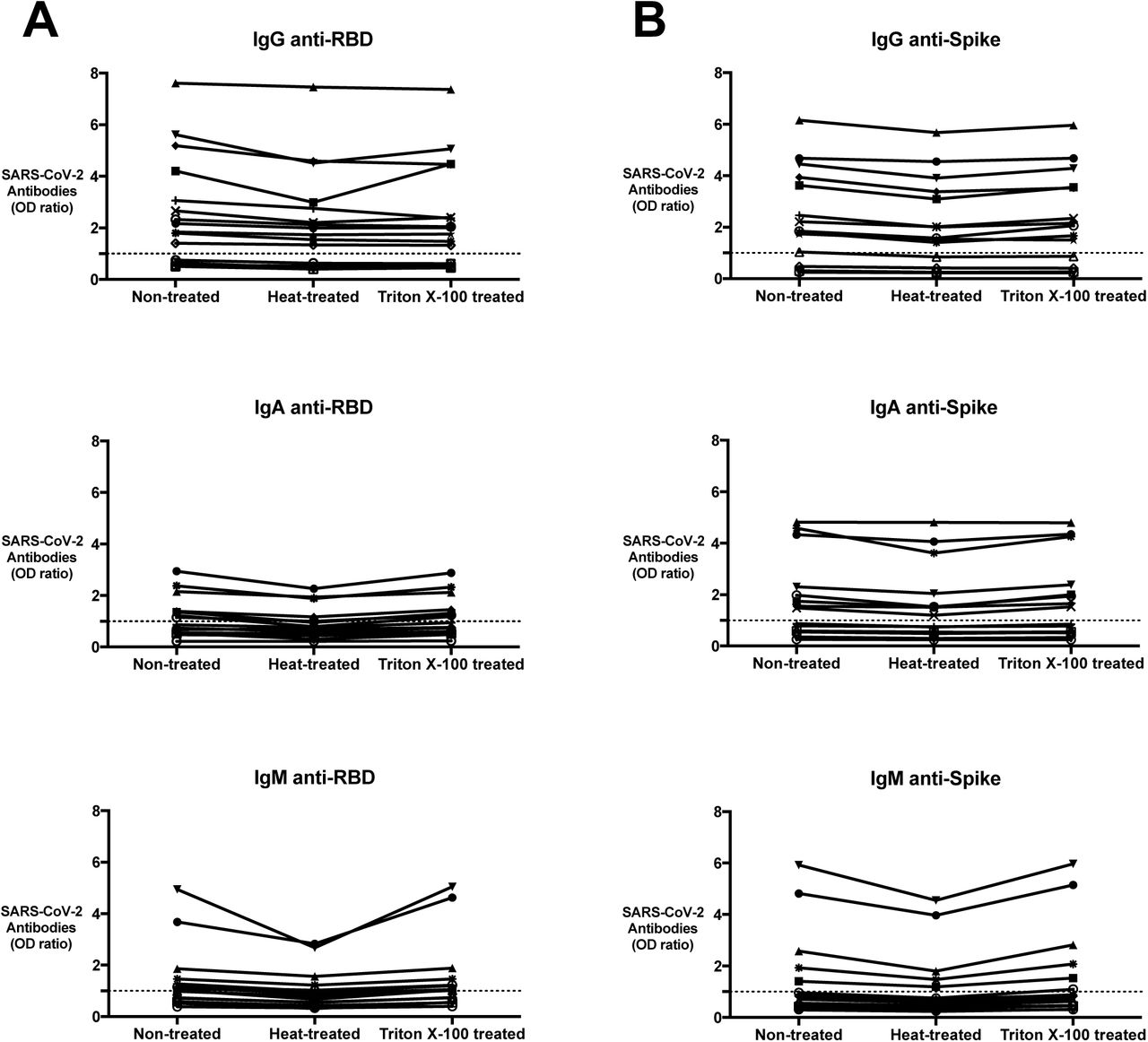
Reactivity of (A) anti-RBD and (B) anti-S protein IgG, A, and M after heat-treatment or Triton X-100 treatment was measured using the SARS-CoV-2 ELISA. Serum samples used for this study included recovered COVID-19 positive patient samples (n=10) and pre-COVID-19 controls (n=5). Values are shown as a ratio of determined optical density to the determined assay cut-off optical density. Values above 1 are considered positive in the SARS-CoV-2 ELISA.
Determining inter-assay variability
A subset of COVID-19 positive patient samples (n=4) and pre-COVID-19 controls (n=4) were tested at least 4 times independently to determine reproducibility of the optimized SARS-CoV-2 ELISA. Upon repeat testing, we found that there was minimal inter-assay variability with repeat testing in anti-RBD IgG on average, results deviated by 4.86±3.53% in COVID-19 positive and 7.14±3.03% in pre-COVID-19 controls (Figure 4). This trend was similar for anti-RBD IgA (7.60±3.75% COVID-19 positive; 5.19±3.02% pre-COVID-19 controls) and IgM (4.21±2.64% COVID-19 positive; 8.58±4.63% pre-COVID-19 controls) and anti-S protein IgG (6.16±5.70% COVID-19 positive; 8.82±2.43% pre-COVID-19 controls), IgA (3.30±2.92% COVID-19 positive; 12.83±4.17% pre-COVID-19 controls), and IgM (6.08±2.68% COVID-19 positive; 12.37±4.18% pre-COVID-19 controls).

Binding of anti-RBD IgG for a subset of COVID-19-positive (n=4) and pre-COVID-19 controls (n=4) were repeat tested at least 4 consecutive times to determine any variability between assays. Filled black symbols represent recovered COVID-19 positive patients and open symbols represent pre-COVID-19 controls. Values are represented as a mean optical density reading with standard deviation at 405 nm. The samples tested for variability using resulted in similar readings and low standard deviations for all repetitions.
Comparing methods for viral inactivation on reactivity of antibodies
We used a panel of COVID-19 positive (n=10) serum samples and pre-COVID-19 controls (n=5) to determine whether heat-inactivation or adding Triton X-100 to the samples would affect anti-RBD or anti-S protein detection. These conditions are commonly used to inactivate virus that may be present in serum prior to downstream serological assays.
The IgG signals were decreased in 12 of 15 samples (66.7%) by an average 13.54±6.73% after heat-treatment, but none of these samples decreased below the cut-off. Signals for IgA against RBD and S protein decreased in 12 of 15 samples (80.0%) by an average 13.85±9.55% after heat-treatment. With heat treatment, for IgM anti-RBD (Figure 3A) and IgM anti-S protein (Figure 3B) from COVID-19 positive patients and pre-COVID-19 controls, there was a reduction in OD by an average 21.0±8.06% after heat-treatment. Four of 10 COVID-19 positive samples IgM ODs decreased below the assay cut-off after heat-treatment.
IgG signals in anti-RBD and anti-S protein were decreased in 14 of 15 samples by an average level of 8.03±7.59% after treatment with Triton X-100. ODs for IgA against RBD and S protein decreased in 7 of 15 samples by an average 4.14±3.38% and increased in the 8 other samples by an average level of 6.74±4.01% after treatment with Triton X-100. With Triton X-100 treatment, IgM anti-RBD (Figure 3A) and IgM anti-S protein had an average increase in OD by 6.4±5.64% in 11 of 15 samples and OD decreased by 3.11±2.68% in 4 of 15 samples. Treatment with Triton X-100 had minimal effect on the reactivity of the samples. None of the COVID-19 positive samples tested with Triton X-100 treatment decreased below the cut-off after treatment. None of the measured pre-COVID-19 controls became higher than the assay cut-off value after heat-inactivation or treatment with Triton X-100.
Inhibition of IgG anti-RBD binding
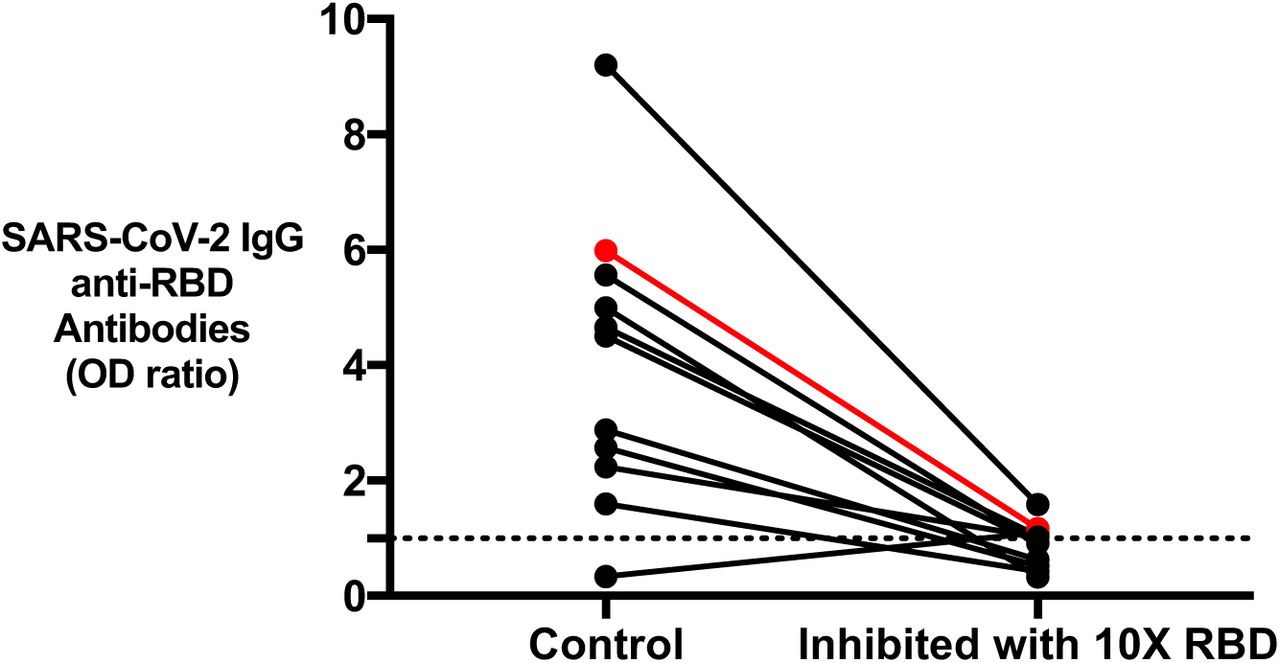
To ensure the detectability of antibody binding in the sera is specific to the antigen and not non-specific binding, the reactivity of COVID-19 positive patient samples (n=10) and 1 antibody-positive sample from pre-COVID-19 controls for anti-RBD IgG was inhibited using excess RBD in solution. All COVID-19 positive patient anti-RBD binding was inhibited on average 78.73±10.12% using excess RBD (Figure 5). One pre-COVID-19 control who tested positive for an anti-RBD IgG was also inhibited to a similar degree (80.51%) as COVID-19 positive samples with excess RBD in solution.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Binding of IgG anti-RBD after inhibition with excess RBD in solution to show the specificity of antibodies to RBD antigen in the SARS-CoV-2 ELISA. Black lines indicate recovered COVID-19 patient samples tested (n=10) and red line represent one antibody-positive pre-COVID-19 control tested. The one pre-COVID-19 control tested positive for IgG anti-RBD antibodies that was also inhibited using excess RBD.
Validation of the SARS-CoV-2 ELISA
We tested a subset of COVID-19 positive (n=9) and negative (n= 5) patient samples in the commercially available EUROIMMUN and the Ortho Clinical Diagnostics assays and compared the results to our in-house ELISA. The EUROIMMUN Anti-SARS-CoV-2 ELISA measures IgG or IgA antibodies to the S1 protein. The Ortho Clinical Diagnostics COVID-19 IgG Antibody Test measures IgG antibodies to the S protein. We found that the in-house SARS-CoV-2 ELISA agreed with the Ortho assay performed by the clinical laboratory (HLRMP), in 14/14 samples (Table 2) for IgG antibodies to the S protein. In one COVID-negative patient sample, the in-house assay detected weak IgG anti-RBD antibodies only, and no other reactivity, which correlated with the Ortho test. The EUROIMMUN assay detected SARS-CoV-2 antibodies in two COVID-19 positive patient samples where the in-house ELISA and Ortho assay did not, one of which was borderline positive for IgG anti-S1.
Discussion
COVID-19 can be a severe infectious disease, however, many patients will have few or no symptoms [31]. Molecular assays to detect the viral genetic material are available for the diagnosis of acute infection, there is a need for accurate serological assays suitable to specifically detect SARS-CoV-2 antibodies [11, 12]. In this study, we describe a high throughput, reproducible, semi-quantitative serological method to detect antibodies against SARS-CoV-2. This assay measures IgG, IgA and IgM antibody-reactivity to the immunogenic S protein and the RBD of the virus. Early serological assessments have revealed some details concerning the antibody responses to COVID-19. Following exposure, patients have an increase in IgG, IgA and IgM with peak IgA and IgM levels at 7-20 days post-infection, followed by IgG [19, 32]. Therefore, by measuring IgG, IgA, and IgM isotypes, we are capturing antibodies elicited at all stages of the immune response and are less likely to encounter false negatives. Furthermore, our in-house ELISA is simple, rapid, does not require specialized instrumentation and can be performed in standard diagnostic laboratories. Reproducibility is one of the factors to consider when establishing an assay. Using a subset of patient samples, we show that after 4 repeated measurements, there is minimal inter-assay variability.
Current COVID-19 antibody tests have shown significant variability [33-35]. Specificity is affected by various factors, including the type of test and the antigen(s). Our assay used the full-length S protein and RBD; however, some groups have tested for antibodies against the nucleocapsid protein of SARS-CoV-2 as well as testing different antibody isotypes (Ortho and EUROIMMUN assays) [36, 37]. Cross-reactivity of antibodies with multiple coronaviruses is an important consideration in developing the SARS-CoV-2 ELISA [38]. Similar to our results, commercial kits of SARS-CoV-2 ELISAs have identified cross-reactivity to presumably seasonal coronaviruses with IgA antibodies having a lower specificity than tests for IgG antibodies [33, 39, 40]. This may affect test specificity as individuals previously infected with seasonal coronaviruses can appear as positives in certain COVID-19 serological tests [41, 42]. In addition, other host factors, including rheumatoid factor and heterophile antibodies, can potentially cross-react and generate false-positive SARS-CoV-2 serology results [43, 44]. These factors may explain why we found 19 pre-COVID-19 controls who tested positive in the SARS-CoV-2 antibody tests. The pre-COVID-19 controls that had anti-RBD antibody were not false positive per se because antibody binding could be inhibited by excess RBD. The most likely explanation for this is that these are cross-reactive antibodies generated to seasonal coronaviruses.
Test specificity is also dependent on the assay cut-off based on the control population tested. We used a biobank containing pre-COVID-19 controls (n=332) and have used these to determine the cut-off of the SARS-CoV-2 ELISA to identify individuals with IgG, IgA and IgM antibodies against RBD or S protein. The optimized ELISA described has a specificity of greater than 94% depending on the antigen and antibody class measured (Table 1). Since our pre-COVID-19 controls include healthy individuals (n=26) and patients suspected of ITP or HIT (n=306), the specificity may not be representative of the general population, but include important hospitalized and elderly patients, potentially exposed to other seasonal coronaviruses. In addition, our in-house ELISA is also in agreement with two commercial assays that report sensitivity and specificity to be 100% and 90%, respectively; however, in some studies testing different populations, sensitivity and specificity of the commercial assays can be as low as 65% and 73.8%, respectively [45, 46].
To date, there have been a number of methods used for virus inactivation. However, how these inactivation methods affect the sensitivity of serological assays is unknown. To reduce risk of exposure to the virus during testing, viral inactivation is recommended before sample handling. We found that with heat-inactivation, detection of serum IgG and IgM was diminished, similar to previous studies [47]. In contrast, Triton X-100 had minimal effect on the antibody reactivity, suggesting that Triton X-100 would be a superior method for virus inactivation as it does not reduce antibody binding in IgM. The decrease in antibody levels may be related to the structural change with denaturation and aggregation when samples are heated [48, 49]. Previous studies have shown that antibodies can lose their antigen binding ability after heating, and IgM is less thermally stable than IgG due to the structure of its heavy chains [50]. Our results indicate that between the two methods, using Triton X-100 for virus inactivation is less likely to affect antibody reactivity.
We describe a high throughput ELISA that detects anti-SARS-CoV-2 antibodies important for identifying COVID-19. This SARS-CoV-2 ELISA described is sensitive, specific and reproducible allowing it to be useful for the investigations of changing infection rates and longitudinal immune responses to SARS-CoV-2. It will be useful to determine antibody titer as it may play a role in determining antibody response with great accuracy and describe disease severity. This will complement RT-PCR tests and help provide evidence of recent infection. The assay could be used to identify antibody responses in patients recovering from COVID-19 disease for convalescent plasma studies and to screen health care workers to allow for selective deployment of personnel to care for different COVID-19 populations. This assay can be used to help with additional longitudinal serological studies profiling symptomatic and asymptomatic individuals to determine the duration of COVID-19 antibody-mediated immunity.
AUTHORSHIP CONTRIBUTIONS
AH carried out the described studies, analyzed data, and wrote the manuscript. DMA designed the research and helped write the manuscript. JWS carried out the described studies, analyzed data, and wrote the manuscript. JCM assisted with experimentation, provided technical assistance, and helped write the manuscript. VTC provided technical assistance. HDS and JCA provided technical assistance and materials. ZG, BH, DMEB, MSM, and JGK designed the research and helped write the manuscript. IN designed the research, interpreted data and wrote the manuscript. All authors reviewed and approved the final version of the manuscript.
DISCLOSURE OF CONFLICTS OF INTEREST
The authors declare no competing financial interests.
ACKNOWLEDGEMENTS
We thank Erjona Kruja for technical assistance. Funding support for this work was provided by a grant from the Ontario Research Fund (ORF), COVID-19 Rapid Research Fund awarded to Dr. Ishac Nazy (#C-191-2426729-NAZY) and Academic Health Sciences Organization (HAHSO) grant awarded to Dr. Donald M Arnold (#HAH-21-02). This work was also supported, in part, by a Weston Family Microbiome Initiative Grant and a Canadian Institutes of Health Research (CIHR) COVID-19 Rapid Response grant to Dr. Matthew S Miller. Dr. Miller was also supported, in part, by a CIHR New Investigator Award and an Ontario Early Researcher Award. Hannah D. Stacey was supported in part by an Ontario Graduate Scholarship.
References




Subject Area
Reviews and Context
0
Comment
0
TRIP Peer Reviews
0
Community Reviews
0
Automated Services
1
Blogs/Media
Author Videos