
Abstract
Background Polygenic hazard scores (PHS) can identify individuals with increased risk of prostate cancer. We estimated the benefit of additional SNPs on performance of a previously validated PHS (PHS46).
Materials and Method 180 SNPs, shown to be previously associated with prostate cancer, were used to develop a PHS model in men with European ancestry. A machine-learning approach, LASSO-regularized Cox regression, was used to select SNPs and to estimate their coefficients in the training set (75,596 men). Performance of the resulting model was evaluated in the testing/validation set (6,411 men) with two metrics: (1) hazard ratios (HRs) and (2) positive predictive value (PPV) of prostate-specific antigen (PSA) testing. HRs were estimated between individuals with PHS in the top 5% to those in the middle 40% (HR95/50), top 20% to bottom 20% (HR80/20), and bottom 20% to middle 40% (HR20/50). PPV was calculated for the top 20% (PPV80) and top 5% (PPV95) of PHS as the fraction of individuals with elevated PSA that were diagnosed with clinically significant prostate cancer on biopsy.
Results 166 SNPs had non-zero coefficients in the Cox model (PHS166). All HR metrics showed significant improvements for PHS166 compared to PHS46: HR95/50 increased from 3.72 to 5.09, HR80/20 increased from 6.12 to 9.45, and HR20/50 decreased from 0.41 to 0.34. By contrast, no significant differences were observed in PPV of PSA testing for clinically significant prostate cancer.
Conclusion Incorporating 120 additional SNPs (PHS166 vs PHS46) significantly improved HRs for prostate cancer, while PPV of PSA testing remained the same.
Introduction
Optimal prostate cancer screening strategies seek to strike a balance between identifying clinically significant and potentially lethal cases that require treatment, while minimizing overdiagnosis of indolent, lower-risk cases that do not need radical treatment1-3. Genetic risk models have emerged as potentially useful tools that identify individuals with greater risk for being diagnosied with prostate cancer4,5, and so help inform if and when to initiate screening for an individual. A subset of these models called polygenic hazard scores (PHS) seeks to directly identify associations between common genetic variants and the age of diagnosis of prostate cancer by utilizing the framework of time-to-event analyses1,6.
We have previously reported on a PHS model for prostate cancer, PHS46, that demonstrated excellent performance in an independent test set of men from varied genetic ancestries6. The model incorporates genetic data of 46 unique single nucleotide polymorphisms (SNPs), and was identified through a systematic search of European men genotyped on the iCOGS chipset (Illumina, San Diego, CA). With an ever-increasing list of loci associated with prostate cancer in the literature7-9, we sought to determine what effect, if any, the incorporation of additional SNPs would have on the performance of PHS46.
To this end, we employed a machine-learning approach, LASSO-regularized Cox regression,10,11 to select SNPs from a list that included the 46 used in PHS46, as well as over 100 SNPs identified in previous analyses as having genome-wide significance for association with prostate cancer7. LASSO-regularized regression is an established variable selection technique in datasets with a large number of predictors and has been previously implemented as a SNP selection tool for a breast cancer polygenic risk score12. Performance metrics describing statistical model goodness-of-fit and clinically actionable screening utility of the LASSO-regularized PHS model for prostate cancer were compared with those achieved with PHS46 to determine the potential benefit of incorporating additional SNPs in polygenic hazard models.
Material and Methods
Study dataset
We obtained genotype and phenotype data from the PRACTICAL13 consortium for this analysis. Genotyping was performed previously on either OncoArray13 or iCOGS9 chips, and these data were previously imputed using the 1000 Genomes reference panel14. Missing SNP calls were replaced with the mean of the genotyped data for that SNP in the training set1,15. In total, data from 82,007 men with European genetic ancestry (Supplementary Table 1)13,16 were available for this analysis. A testing set consisting of 6,411 men (4,828 controls and 1,583 cases) enrolled in the ProtecT clinical trial was set aside for estimating the performance of the final PHS models. The data from ProtecT were chosen as the testing set because they are well characterized and were previously used for validation of PHS461, allowing us to directly benchmark the performance of the updated model against previous iterations. The ProtecT trial also included biopsies of participants with elevated prostate-specific antigen (PSA) level, which permits analysis of the positive predictive value of the current clinical standard for screening, PSA testing. The remaining 75,596 individuals (25,127 controls and 50,469 cases) were used for training of the model. This first analysis was limited to men of European descent because of much greater data availability in that population, but our previous work has shown that development in Europeans can inform careful future work to assess and improve performance in other ancestries17.
Model development using LASSO regularization
A list of published SNPs previously identified1,7 to be associated with prostate cancer was compiled. In total, 180 unique SNPs were considered for estimation within the PHS model framework. An initial screening was conducted to identify pairs of SNPs that were highly correlated (R2 > 0.95). For each pair of highly correlated SNPs, a univariable Cox proportional hazards model using age of diagnosis of prostate cancer as the time to event was calculated for each SNP in the pair, and the one with the larger p-value was discarded. The remaining SNPs were included as candidates for the new PHS model. The R package ’glmnet’ was used to estimate a LASSO-regularized Cox-proportional hazards model10,11 using age of diagnosis of prostate cancer as the time to event. The genetic data of candidate SNPs and first four European ancestry principal components were included as predictors. Controls were censored at age of last follow-up. The hyper-parameter of the LASSO-regularized model, lambda, was selected using 10-fold cross-validation10,11. The final form of the LASSO model was estimated at the value of lambda that minimized the mean cross-validated error.
Characterization of LASSO-regularized PHS model
The PHS score for each of the individuals in the training and testing set was estimated as the weighted sum of the genetic counts of each of the SNPs in the PHS model, using the LASSO model coefficients as weights. Distributions of the new PHS score were compared qualitatively between training and testing groups to confirm that the model was appropriately calibrated for use in the testing set.
Performance comparison between PHS46 and LASSO-regularized PHS
Performance in the testing set was assessed using hazard ratios (HRs) and positive predictive value (PPV), as described below. In each case, performance metrics were generated for the newly developed LASSO PHS model and for PHS46. Model coefficients for PHS46 were obtained from the literature17. For each performance metric, one thousand bootstrap samples of the testing set were used to generate empirical 95% confidence intervals for LASSO PHS and for PHS46. In addition, bootstrapped 95% confidence intervals were generated for the percentage change of each performance metric between the two models, using PHS46 as the 25 reference. Percent changes were deemed statistically significant if the bootstrapped 95% confidence interval did not include 0.
HR performance
Calibration Cox proportional hazards models were fit to the bootstrapped testing data using the PHS score as the sole predictor and the age-of-diagnosis of prostate cancer as the dependent variable. The model coefficient of this Cox regression model is referred to as the calibration factor. Next, the hazard ratio between two PHS groups, such as those in the top 5% to the middle 40% (HR95/50), is estimated as the exponential of the product of the calibration factor and the difference in mean PHS scores of each group. Hazard ratios between the top 20% to the bottom 20% (HR80/20) and the bottom 20% to the middle 40% (HR20/50) were similarly calculated. The PHS cutoffs used to define these groups were determined from the distribution of PHS in the training set controls under 70 years of age1,15.
A similar strategy was used to estimate the HR performance for clinically significant prostate cancer. The criteria for clinical significance were any of: Gleason score >=7, stage T3-T4, PSA concentration >= 10ng/mL, pelvic lymph nodal metastasis, or distant metastasis18. In this analysis, controls and low-risk (i.e., not clinically significant) cancers were censored at age of last follow-up and age of diagnosis, respectively. HRs are reported after sample-weight correction1,17,19 using the total number of cases and controls in the ProtecT trial to generate weighting factors.
PPV performance
One indicator of clinical utility of a risk-stratification approach like PHS is whether it can be used to improve the PPV of the standard clinical screening test, prostate-specific antigen (PSA). As a population-based screening study, ProtecT provides biopsy results of both cases and controls with a positive PSA result (i.e., ≥/mL). PPV performance of each model was estimated by randomly sampling individuals within the testing set with positive PSA results, while maintaining the case to control ratio of the ProtecT study (1:2). PPV is calculated as the fraction of positive PSA individuals in the top 20% (PPV80) or top 5% (PPV95) of PHS scores that had clinically significant prostate cancer.
Cumulative incidence curves for LASSO-PHS in United Kingdom
To illustrate the utility of the LASSO PHS model in informing prostate cancer screening, cumulative incidence curves for various PHS risk groups were estimated, as described previously20. The age-specific general cumulative incidence curve for prostate cancer was estimated for the United Kingdom population, aged 40 to 70, using data from Cancer Research UK 2015-201721. The proportion of clinically significant and non-clinically significant prostate cancer at each age was estimated using data from the Cluster Randomized Trial of PSA Testing for Prostate Cancer (CAP) trial22. Disease-specific cumulative incidence curves for clinically significant and non-clinically-significant prostate cancer were estimated by multiplying the general cumulative incidence curve by their respective proportions. The risk-adjusted incidence curves for individuals in the upper 5th percentile and upper 20th percentile were estimated by multiplying the disease-specific cumulative incidence curves by the mean value of HR95/50 and HR80/50 in the testing set, respectively. Hazard ratios were obtained using the age of diagnosis of clinically significant prostate cancer as the time-to-event and after sample-weight correction.
Results
SNP screening and PHS model training
Of the 180 SNPs originally considered for this study, 6 SNPs were discarded in the initial screening process of removing highly correlated SNPs. Of the 174 remaining candidate SNPs (Supplementary Table 2), 166 had non-zero LASSO model coefficients and were selected for the final PHS model (PHS166).
PHS166 model characterization
Distributions of PHS166 score were visually consistent between training and testing sets (Supplementary Figure 1). The 20th, 30th, 70th, 80th, and 98th percentiles of the reference PHS risk scores (controls in training set) were estimated as -0.411, -0.307, 0.048, 0.154, and 0.557, respectively.
Performance comparison –PHS46 vs. PHS166
All PHS166 HR-based performance metrics showed statistically significant improvements compared to PHS46 (Table 1), for both any and clinically significant prostate cancer. The mean HR95/50 and HR80/20 values for PHS166 were roughly 36 to 55% greater than those for PHS46. For example, HR80/20 for clinically significant prostate cancer increased from 6.12 to 9.45. Similarly, HR20/50 for PHS166 was, on average, 18% lower than that for PHS46. No significant differences between models were observed in either of the PPV-based performance metrics (Table 2). Among individuals in the top 20% of risk scores with a positive PSA test, the estimated mean PPV for clinically significant prostate cancer was roughly 0.19 irrespective of the model used – indicating approximately 19% of positive PSA tests in this risk group yielded a diagnosis of clinically significant prostate cancer. By comparison, approximately 13% of all positive PSA tests resulted in a diagnosis of clinically significant prostate cancer.
Sample-weight-corrected hazard ratios are estimated for PHS166 and PHS46 in the testing set, using age-of-onset of any or clinically significant prostate cancer. The percent change for each metric is calculated using the value of PHS46 as the reference. Mean values and 95% confidence intervals are reported.
Positive predictive value (PPV) of PSA testing for clinically significant prostate cancer using top 5% (PPV95) and top 20% (PPV80) cutoffs of PHS166 and PHS46 risk scores. The percent change for each metric is calculated using the value of PHS46 as the reference.
Cumulative incidence curves for PHS166 in United Kingdom
Cumulative incidence curves for clinically significant and non-clinically significant prostate cancer for the upper 5th percentile (>95th percentile) and upper 20th percentile (>80th percentile) of PHS166 scores in the United Kingdom demonstrated expected stratification of prostate cancer risk (Figure 1).
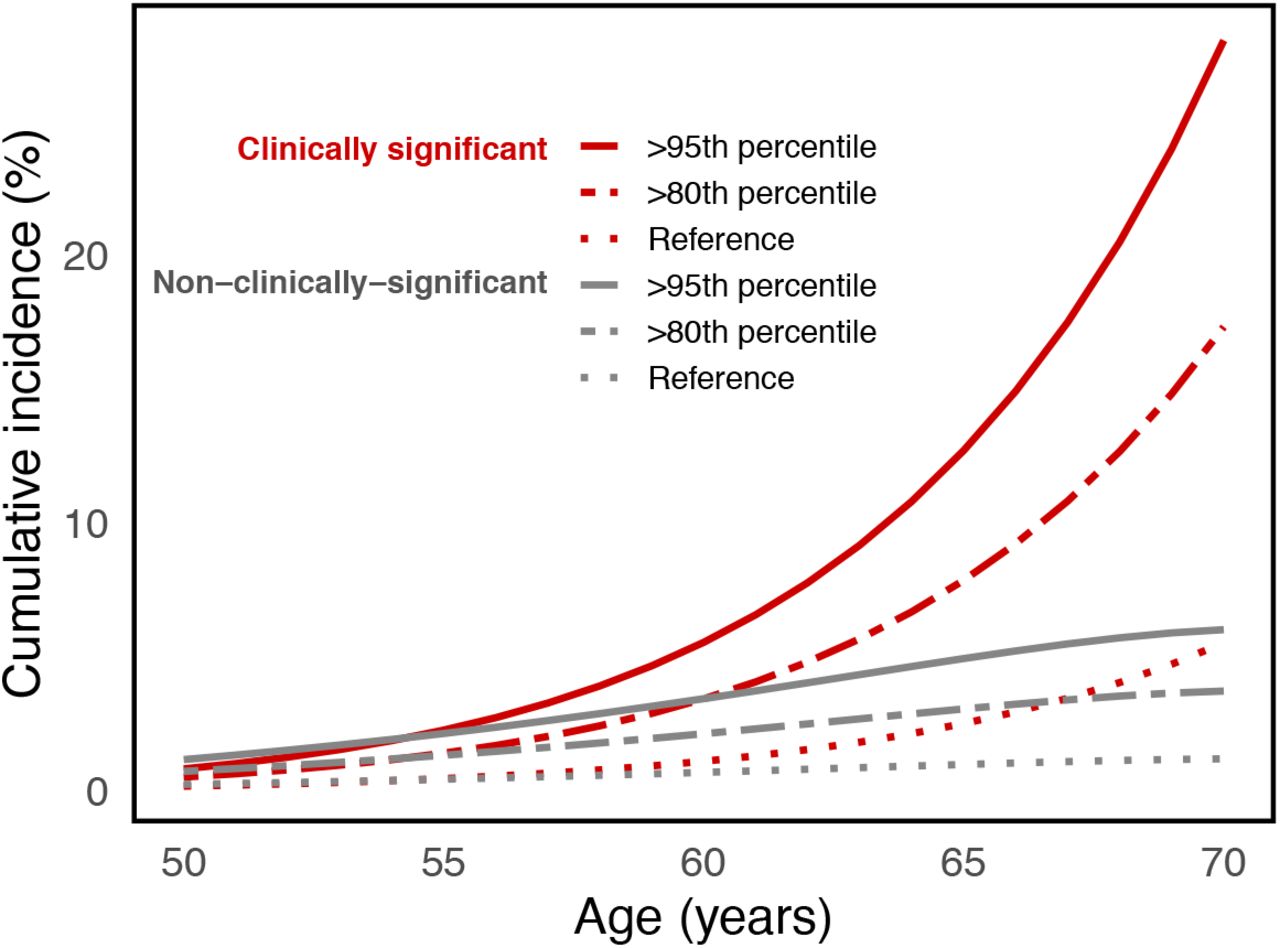
{kind=link}
Risk-adjusted cumulative incidence curves for the upper 5th percentile (>95th percentile) and upper 20th percentile (>80th percentile) of PHS166 scores for clinically significant and non-clinically-significant prostate cancer. Reference curves representing the population average cumulative incidence (i.e., unadjusted for genetic risk).
Discussion
Using a machine-learning, LASSO-regularized Cox framework, we identified 166 SNPs to be included in a polygenic hazard model (PHS166) for association with age of diagnosis of prostate cancer in men of European genetic ancestry. When compared to the original PHS, consisting of 46 SNPs, PHS166 demonstrated substantially improved HR performance. For example, the HR for clinically significant prostate cancer comparing the upper and lower quintiles of genetic risk increased by 56% when using PHS166. No significant improvements were found in the PPV of PSA testing when using PHS to stratify risk.
Increased separation in hazard rates between PHS risk groups may allow for more nuance in clinical decision making in certain scenarios. Accurate identification of low, intermediate, and 10 high PHS risk groups in prostate cancer may help in decisions of when (or if) to initiate screening as well as possibly improving the interpretation of the disease screens23. Targeting screening to men in the upper percentiles of polygenic risk as opposed to those in the lowest risk group may reduce the proportion of overdiagnosed indolent cancers from 43% to 19%24,25. Risk stratification achieved here by PHS166 is similar or better than commonly used clinical tools for diseases such as breast cancer, diabetes, and cardiovascular disease23,26-28. Clinically meaningful risk stratification is illustrated by the estimated cumulative incidence curves in Figure 1. This effect is particularly pronounced for clinically significant disease because of the increased proportion of clinically significant cases observed at older ages2,20,22.
The lack of improvement in PPV in this study may suggest a “performance plateau” when using PHS to define broad risk categories for certain clinical applications. A similar effect has been previously described for prostate cancer polygenic models, in the context of using risk scores to discriminate prostate biopsy outcomes29. Some of the precision in a score may also be diluted in broad clinical applications. The PPV analysis here is applied to participants in the ProtecT trial, which enrolled men aged 50 to 69 years, and screening in the trial was offered irrespective of underlying genetic risk2. Further investigation is needed to learn whether timing screening according to genetic risk might better leverage the superior HR performance of PHS166 risk score to improve the PPV of PSA testing.
LASSO frameworks have been used to identify SNPs for polygenic risk scores of several phenotypes, including fracture risk30, type 2 diabetes31, and breast cancer12. In this work, we have extended the application of LASSO to select SNPs in a polygenic hazard model of prostate cancer from a list of candidates previously identified through logistic and time-to-event analysis. Simulation studies11 have suggested that LASSO provides more robust estimates than stepwise selection in cases with both a few large effects, as well as many small effects. As new prostate cancer associated variants are discovered, this framework can be easily implemented to develop updated polygenic hazard models.
One limitation of PHS166 is that it was entirely developed and tested in European men. However, a well-vetted, well-tested PHS model for men of European genetic ancestry can be used as a starting block for developing models for other genetic ancestries, where large-scale databases are often more scarce, as has been shown for PHS4617,32. Furthermore, some of the SNPs selected for incorporation into PHS166 were originally discovered in analyses that included men from the ProtecT testing set. Therefore, the improvements in HRs observed for PHS166 may be somewhat overestimated. However, this bias is likely small, given that the testing set was only a small fraction of the data used in prior discovery analyses, and the ProtecT data were not used to calculate SNP weights in PHS166. In addition, this study uses age of diagnosis as the time-to-event variable, and any preceding period of undiagnosed disease is unknown. Hypothetical perfect measurement of age of onset would likely further improve performance of the PHS model.
In conclusion, we applied a machine-learning, LASSO-regularized Cox regression framework to develop a larger PHS that includes 166 previously discovered SNPs. When comparing the performance of PHS166 to the original model, PHS46, we found that incorporating 120 more SNPs significantly improved HRs for clinically significant prostate cancer. However, incorporating more SNPs did not improve on the ability of PHS46 to inform the PPV of PSA testing in the ProtecT dataset, perhaps illustrating a plateau effect and/or dilution of risk stratification in a broad clinical application.
Data Availability
The data used in this work were obtained from the Prostate Cancer Association Group to Investigate Cancer Associated Alterations in the Genome (PRACTICAL) consortium. Readers who are interested in accessing the data must first submit a proposal to the Data Access Committee. If the reader is not a member of the consortium, their concept form must be sponsored by a principal investigator (PI) of one of the PRACTICAL consortium member studies. If approved by the Data Access Committee, PIs within the consortium, each of whom retains ownership of their data submitted to the consortium, can then choose to participate in the specific proposal. In addition, portions of the data are available for request from dbGaP (database of Genotypes and Phenotypes) which is maintained by the National Center for Biotechnology Information (NCBI): https://www.ncbi.nlm.nih.gov/gap/?term=Icogs+prostatehttps://www.ncbi.nlm.nih.gov/gap/?term=Icogs+prostate. Anyone can apply to join the consortium. The eligibility requirements are listed here: http://practical.icr.ac.uk/blog/?page_id=9. Joining the consortium would not guarantee access, as a proposal for access would still be submitted to the Data Access Committee, but there would be no need for a separate member sponsor. Readers may find information about application by using the contact information below: Rosalind Eeles Principal Investigator for PRACTICAL Professor of Oncogenetics Institute of Cancer Research (ICR) Sutton, UK Email: PRACTICAL@icr.ac.uk URL: http://practical.icr.ac.uk Tel: ++44 (0)20 8722 4094
Ethics Statement
All contributing studies were approved by the relevant ethics committees and performed in accordance with the Declaration of Helsinki; written informed consent was obtained from the study participants. The present analyses used de-identified data from the PRACTICAL consortium and have been approved by the review board at the corresponding authors’ institution.
Conflict of Interest
All authors declare no personal or financial conflicts of interest for the submitted work except as follows. CCF is a scientific consultant for CorTechs Labs, Inc. RE reports honorarium as a speaker for GU-ASCO meeting in San Francisco Jan 2016, support from Janssen, and honorarium as speaker for RMH-FR meeting Nov 2017. She reports honorarium as a speaker at the University of Chicago invited talk May 2018, and an educational honorarium by Bayer & Ipsen to attend GU Connect “Treatment sequencing for mCRPC patients within the changing landscape of mHSPC” at ESMO Barcelona, Sep 2019. She reports member of external Expert Committee on the Prostate Dx Advisory Panel. OAA received speaker’s honorarium from Lundbeck, and is a consultant for Healthlytix. AMD reports that he was a founder and holds equity in CorTechs Labs Inc., and serves on its Scientific Advisory Board. He is a member of the Scientific Advisory Board of Human Longevity, Inc., and the Mohn Medical Imaging and Visualization Centre. He received funding through research grants from GE Healthcare to UCSD. The terms of these arrangements have been reviewed by and approved by UCSD in accordance with its conflict of interest policies. TMS reports honoraria, outside of the present work, from: University of Rochester, Varian Medical Systems, Multimodal Imaging Servcies Corporation; and WebMD. He reports research funding from NIH/NBIB, U.S. Department of Defense, Radiological Society of North America, American Society for Radiation Oncology, and Varian Medical Systems.
Data Availability Statement
The data used in this work were obtained from the Prostate Cancer Association Group to Investigate Cancer Associated Alterations in the Genome (PRACTICAL) consortium. Readers who are interested in accessing the data must first submit a proposal to the Data Access Committee. If the reader is not a member of the consortium, their concept form must be sponsored by a principal investigator (PI) of one of the PRACTICAL consortium member studies. If approved by the Data Access Committee, PIs within the consortium, each of whom retains ownership of their data submitted to the consortium, can then choose to participate in the specific proposal. In addition, portions of the data are available for request from dbGaP 10 (database of Genotypes and Phenotypes) which is maintained by the National Center for Biotechnology Information (NCBI): https://www.ncbi.nlm.nih.gov/gap/?term=Icogs+prostatehttps://www.ncbi.nlm.nih.gov/gap/?term=Icogs+prostate.
Anyone can apply to join the consortium. The eligibility requirements are listed here: http://practical.icr.ac.uk/blog/?page_id=9. Joining the consortium would not guarantee access, as a proposal for access would still be submitted to the Data Access Committee, but there would be no need for a separate member sponsor. Readers may find information about application by using the contact information below:
Rosalind Eeles
Principal Investigator for PRACTICAL Professor of Oncogenetics Institute of Cancer Research (ICR) Sutton, UK Email: PRACTICAL{at}icr.ac.uk URL: http://practical.icr.ac.uk Tel: ++44 (0)20 8722 4094
Funding
This study was funded in part by a grant from the United States National Institute of 5 Health/National Institute of Biomedical Imaging and Bioengineering (#K08EB026503), the Research Council of Norway (#223273), KG Jebsen Stiftelsen, and South East Norway Health Authority.
References



