
Summary
In February 2020 a substitution at the interface between SARS-CoV-2 Spike protein subunits, Spike D614G, was observed in public databases. The Spike 614G variant subsequently increased in frequency in many locations throughout the world. Global patterns of dispersal of Spike 614G are suggestive of a selective advantage of this variant, however the origin of Spike 614G is associated with early colonization events in Europe and subsequent radiations to the rest of the world. Increasing frequency of 614G may therefore be due to a random founder effect. We investigate the hypothesis for positive selection of Spike 614G at the level of an individual country, the United Kingdom, using more than 25,000 whole genome SARS-CoV-2 sequences collected by COVID-19 Genomics UK Consortium. Using phylogenetic analysis, we identify Spike 614G and 614D clades with unique origins in the UK and from these we extrapolate and compare growth rates of co-circulating transmission clusters. We find that Spike 614G clusters are introduced in the UK later on average than 614D clusters and grow to larger size after adjusting for time of introduction. Phylodynamic analysis does not show a significant increase in growth rates for clusters with the 614G variant, but population genetic modelling indicates that 614G increases in frequency relative to 614D in a manner consistent with a selective advantage. We also investigate the potential influence of Spike 614D versus G on virulence by matching a subset of records to clinical data on patient outcomes. We do not find any indication that patients infected with the Spike 614G variant have higher COVID-19 mortality, but younger patients have slightly increased odds of 614G carriage. Despite the availability of a very large data set, well represented by both Spike 614 variants, not all approaches showed a conclusive signal of higher transmission rate for 614G, but significant differences in growth, size, and composition of these lineages indicate a need for continued study.
Introduction
A recurring concern during recent viral epidemics has been whether mutations will arise that change the fundamental properties of the virus, such as its mode or rate of transmission, or its ability to cause disease. While fast-evolving RNA viruses will continually exhibit new mutations throughout their genomes and whilst most will be effectively neutral, identifying those that are not is challenging and predicting effect of these on the fitness of the virus is generally not possible, particularly in a newly emerged virus like SARS-CoV-2. Furthermore, the epidemiological processes driving the pandemic – the spread of the virus from one region to another, the seeding of new epidemics in susceptible populations – will amplify any mutations that happen to be carried by these viruses whether or not they confer any selective advantage.
In late February, a non-synonymous mutation in the SARS-CoV-2 spike (S) protein - a change from aspartic acid to glycine at position 614 of the S protein (D614G) - began to be noticed in viruses sampled from individuals from China and Europe.The S protein, extensively studied in other coronaviruses including SARS-CoV[1–3] and MERS[4,5], is a glycoprotein critical to cell entry and as such mutations in this gene have the potential to change the biology of the virus within a host, for example, altering binding affinity to ACE2 and, as a consequence, transmissibility.
D614G is strongly associated with the B.1 lineage (Figure 1), which now dominates the global pandemic (https://cov-lineages.org/lineages/lineage_B.1.html), and retrospectively sampled viruses place this mutation in Guangzhou, Sichuan and Shanghai Provinces, China in late January (Figure S1). In Europe, the 614G variant was first sampled on January 28th in a small outbreak in Bavaria, Germany, which was seeded by a visitor from Shanghai[6] and through public-health efforts the introduction was controlled. It is therefore likely that the 614G variant arose in China before seeding, on multiple occasions, the large outbreaks in Italy and Central Europe[7]. In the UK, the first sampled virus carrying the 614G variant was from the 28th of February in Scotland from a patient who had recently travelled through Italy [8]. This report is consistent with the rapid expansion of viruses carrying the 614G variant in Europe being submitted to GISAID in February and March [9,10].

Maximum likelihood phylogeny reconstructed from SARS-CoV-2 sequences showing global lineage assignment and the origins of the Spike 614G clade which seeded many introductions in the United Kingdom. Putative reversions to 614D and independently arising D614G mutations are shown as large circles. A number of D614N genomes are shown as red circles, comprising two independent clusters in the UK.
Three distinct sets of observations form the basis for the putative importance of D614G in the SARS-CoV-2 pandemic. Firstly, experimental work using pseudotyped lentiviruses demonstrate that the D614G replacement increases infectivity in vitro [9,11,12]. Secondly, structural analysis indicates that D614G alters the RBD conformation such that ACE2 binding and fusion have higher probability [12]. Finally, analysis of the frequency of the 614D and 614G variants over time (based on submissions to global sequence databases) have suggested that locations that recorded 614D viruses early in the pandemic, were generally later dominated by 614G viruses [9,13].
There is currently no scientific consensus on the effect of the D614G mutation on SARS-CoV-2 infections, and skepticism that it could produce a meaningful effect at the population-level in an already highly-transmissible and rapidly spreading virus [14,15]. Evaluating the in vitro effect of such mutations on the infectivity of pseudotyped viruses carrying the SARS CoV-2 spike protein is an important first step but determining whether such observations hold true in complete SARS-CoV-2 variants in vitro is more challenging, and ultimately requires an animal model that effectively reproduces meaningful aspects of virus infection and transmission. There are also limitations with translating observations from experimental systems to human infections. Therefore detecting changes in human to human transmission requires large-scale population-wide sequencing studies. The relatively small size of many countries’ SARS-CoV-2 genomic datasets precludes robust analysis on a country scale in most cases. The substantially larger global SARS-CoV-2 sequence dataset is problematic because of limited sequence metadata and variable sampling approaches between countries. To statistically determine if there is a meaningful difference between the 614 variants, we need to identify repeated independent introductions of each variant into the same population and follow the trajectories of the outbreaks they cause.
In the UK, the early creation of a national sequencing collaboration, CoG-UK [16], has seen the production of over 30,000 SARS-CoV-2 sequences from across the country in less than 5 months. CoG-UK has enabled the usage of consistent sampling, shared bioinformatic and laboratory approaches, and the collection of the same core set of metadata for all samples. This has produced a substantial, high-resolution dataset that is designed to enable the examination of changes in virus biology in the UK. Crucially, for this study, the UK epidemic is the result of repeated introduction of SARS-CoV-2 from numerous global locations including a substantial number of both 614D and 614G carrying viruses. Here we make use of the CoG-UK SARS-CoV-2 dataset – comprising approximately half of all genomes sequenced globally as of the 7th of July – to examine evidence for increased transmissibility due to changes in the Spike protein.
Results
We identified 21,231 614G and 5,755 614D de-duplicated whole genome sequences sampled from different infections within the UK and with known dates of sample collection collected between January 29 and June 16, 2020. We estimated phylogenetic clusters within the United Kingdom using a maximum-parsimony reconstruction of the location of lineages within the global SARS-CoV-2 phylogeny (c.f. Methods). Each lineage/cluster approximately stems from one or a small number of introductions from outside of the UK. We identify 245 614G and 62 614D clusters with at least 10 different patient samples. Importantly, more lineages with the 614G variant than the 614D variant were introduced into the UK, and these introductions happened later on average (Figure 2). The time of the first sample within 614G clusters is on average 16 days after 614D clusters. Whereas the frequency of sampling of 614G and 614D variants was close to parity early in the epidemic, 614G became the dominant form in late March and this trend has continued (Figure 2C).

Geographic and temporal distribution of Spike 614 amino acid D or replacement G and time of lineage introductions into the United Kingdom. A) Shaded regions show the predominant residue in each region on the 15th of each month for March, April, May and June 2020, with orange indicating that 614G was more sampled and green indicating that 614D was more sampled, or that they were equally sampled. Light grey indicates that no sequences had been sampled by that point in time, and dark grey indicates the Republic of Ireland. B) The time when an introduction is first detected in the United Kingdom for Spike variants 614D and 614G. Singleton introductions are sampled exactly once and not observed subsequently, and lineage introductions gave rise to multiple sampled genomes. Solid lines show the total number of sequences collected by day of each 614 variant. C) The log odds of sampling a Spike 614G variant over time. D) The size of cluster versus time of first sample collected within a cluster.
Evaluating the hypothesis of increased transmission fitness of the 614G variant
Lineages introduced into the UK early in the course of the epidemic and before stringent public health measures were put in place tend to be larger than lineages introduced later (Figure 2D). A large proportion (approximately 50% using accelerated transition parsimony) of samples are descended from several introductions in late January or early February 2020. Although most 614G clusters were introduced later, they are 59% larger on average than 614D clusters after adjusting for time of first sample (p=0.008).
To evaluate if increasing sampling frequency of 614G reflects a selective advantage, we fit a logistic growth model to observed sample collection dates under the assumption that each sample is collected in proportion to the abundance of the 614G variants in the population which changes over time. Under this model, the number of individuals infected with the 614D variant grows exponentially at a rate and the number with the 614G variant grows exponential at rate r (1 + s). The variable represents the selection coefficient which is estimated.
As the increasing frequency of 614G sampling may be an artifact of increasing rate of introductions (founder effects), we adapted the logistic growth model to only count samples which were derived from clusters initiated in January or February. We further limit the analysis to samples collected during a period of exponential growth up to the end of March when a national lockdown was implemented in the UK. Origin times for clusters were estimated using molecular clock phylogenetic methods (c.f. Methods). We also only consider samples collected after the most recent common ancestor (TMRCA) of all clusters and where there are at least 10 samples with either amino acids 614D or G, so that all samples were collected during a period when these clusters were co-circulating within the UK. For this analysis, 5 614D clusters (n=355 sequences) and 5 614G clusters (n=1,855 sequences) were retained. We estimated a selection coefficient of 0.21 (95% CI: 0.06 - 0.56) (Table 1). The observed and fitted frequencies of 614G samples are shown in Figure S2. Information used to fit this model is drawn disproportionately from late March when more samples are available.Some regional variation in transmission rates was discernible as the effect appears stronger including sequences from England (s=0.34, 95%CI:0.13-0.88, n=397), but no effect is observed using data from Scotland (95%CI: -1.78- 1.2, n=60).
Estimates of the selection coefficient favouring the Spike 614G variant using different sources and methods.
We separately fitted the logistic growth model to a period of epidemic decline after April 15. Including all clusters detected before March 31, we have n=3,335 (3,093 614G and 242 614D) samples observed after April 15 descended from 37 clusters. This cross-section of data also shows a similar trend towards increasing frequency of 614G (Figure S2). Adjusting the timescale to match a generation time between 3 and 8 days, we estimated a selection coefficient of 0.27 (95% CI:0.12-0.54).
An alternative source of information about growth rates comes from changing patterns of genetic diversity over time in each cluster. We applied phylodynamic methods[17] to estimate effective population size and effective growth rates over time. First, we applied a parametric ‘boom-bust’ exponential growth coalescent model to all lineages with more than 40 samples, giving 50 lineages (11 containing the D wildtype, 39 containing the G variant).
According to this model, population size grows exponentially up to a transition point which was estimated independently for each cluster, whereupon it shrinks exponentially. Rates of growth and decline are estimated separately for 614G and 614D clusters. Among these 50 clusters, 614G tended to start later and persist longer than 614D (Figure S4), and 614D lineages tended to have slightly earlier transition time (614D mean = 24th March, 614G mean = 2nd April). We do not detect any significant selection effect with this model (Table 1), and there was large uncertainty in cluster growth rates (Figure S5). Growth rates in 614G tended to be larger (posterior mean growth 117 1/year versus 93 1/year). There was not a significant difference between the rate of decline of 614G and 614D clusters (posterior mean -11 1/year versus -9 1/year).
Secondly, we applied a non-parametric phylodynamic model which allows the growth rate of population size to vary over time according to a stochastic process. We applied this model independently to each of the clusters described above. We found that effective population size in the largest clusters tracks the progression of the epidemic in the UK and growth in most clusters is negative by early April 2020 (Figure S6). We then examined if the Spike D614G replacement explained variance in growth rates between transmission clusters. The initial growth rate at the time of MRCA for each cluster was highly variable (Figure 3A) and precision of the estimated rate was generally low (Figure S6). The Spike 614 polymorphism explains very little variance in growth rates between clusters on its own (weighted least squares R2=1%). There was not a significant difference in the initial growth rates of the clusters (Kruskal Wallis p = 0.13). The median initial growth rate among 614D-associated clusters was 117 1/year and among 614G clusters was 169 1/year. This corresponds to an R0 of 3.1(interquartile range (IQR): 2.7-3.5)) for 614D clusters and 4.0(IQR: 3.1-4.8) for 614G clusters if assuming a 6.5 day serial interval[18]. Region of sample collection was not significantly associated with growth rates (weighted least squares, p=0.248). We did not observe a significant association between growth rates and when clusters were first sampled (weighted least squares, p=0.62).

{kind=link}
{kind=link}
{kind=link}
Estimated growth rates, effective infections over time, and estimated versus actual odds of sampling Spike 614G over time. A) Estimated growth rates (1/year) for 48 UK clusters at the estimated TMRCA of each cluster. Size and colour of points indicate the number of samples from each lineage and the country with more than 50% of samples. B) Estimated effective infections for a phylodynamic SEIR model fitted to 200 London sequences and 100 sequences from outside of London. Orange and green show respectively the number of 614G and 614D infections and shaded regions show 95% credible intervals. C) The estimated log odds of sampling 614G over time based on the relative size of 614G and 614D populations shown in panel (B). The shaded region shows the 95% credible interval and the black line shows the empirical log odds of sampling 614G based on a random sample of 1000 sequences. D) As panel (C) and incorporating information about observed odds of sampling 614G over time.
We then examined if there was a detectable difference in growth rates using information from both the phylogeny and the empirical frequency of sampling the 614G variant over time. We conducted a model-based phylodynamic analysis using 200 sequences sampled from the London metropolitan area (c.f. Phylodynamic Methods). A phylogeographic model specified the relationship between the London data and a random sample of 100 sequences from outside of London which provides a mechanism to control for founder effects. Figure 3B shows the estimated number of 614G and 614D infections over time in London. We estimated that 614D was initially the most prevalent variant but that 614G overtook Spike 614D in late March 2020. A similar transition from 614D to 614G was observed in the empirical distribution of samples, such that by the end of March samples from London are more than twice as likely to have the 614G variant. The phylodynamic model was fitted both with and without information about sampling frequency of 614G over time. Incorporating sampling information substantially modifies the estimated selection coefficient upwards from 0.10 to 0.26 (95% CI: −0.01-0.58) (Table 1). It is important to note that all fitted trajectories predict that the log odds of sampling 614G increase even if the selection coefficient is zero and that this is not necessarily evidence of positive selection for 614G.
Association of Spike 614 D versus the G replacement with infection outcome and age
By linking genetic sequence data with clinical data on patient outcomes we were able to investigate associations between viral 614 residue and virulence. We studied 732 614G and 602 614D associated genetic sequences collected by Public Health England between 5 February and 17 March 2020 which could be linked to patient outcome after 28 days post diagnosis (death or recovery). In univariate analyses, patients with the 614G variant show reduced odds of death, but this effect disappeared after controlling for other known risk factors for severe COVID19 outcomes (Table 2). We observed associations between time of sampling and genotype (later samples were more likely to have 614G) and later samples having higher odds of death and higher age. Odds of survival decrease for later samples, which may reflect prioritization of very severe cases for hospitalization and genetic sequencing as the epidemic peaked in March and April.
Odds ratios (OR) of death within 28 days post diagnosis. Continuous variables were scaled (Z score) before regressing. Coefficients are in standardized units. 95% confidence intervals are shown in parentheses.
We observed an association between age and genotype with younger patients more likely to carry 614G viruses. This association is observed despite the progressive aging of the patient cohort and concomitant increase in prevalence of 614G relative to 614D. We performed a multivariate analysis on the metadata of 27,038 sequences from across the UK (England, Wales, Scotland, and Northern Ireland) for the sample collection date, the age and sex of patients. A significant difference was found between the distribution of patient ages for 614G and 614D (Figure S8, Mann Whitney U test: p < 10−13). The median age is 5 years older among female carriers of 614D versus 614G and 4 years older among male carriers of 614D versus 614G. An association was also observed between sex and the presence of 614G or 614D (Figure S8, Chi-squared test: p < 10−10). Differences in the age distribution for each sex were also observed (Mann-Whitney U p<10−8 for 614D and p<10−37 for 614G). The probability of carrying 614G virus seems to decrease continuously with age (Figure S8). Possibly this is due to an increased viral load in younger patients associated with 614G variants leading to higher detection rates.
As a proxy for viral load we studied 12,082 sequences with PCR cycle threshold (Ct) values from across the UK. Sequences with 14≤ Ct ≤ 40 were inspected for association with genotype and a very slight (<1 Ct step) but significant difference was observed with 614G associated with lower Ct (Figure S6, p <10−6, Mann-Whitney U test). Note that different test methods were used to obtain the Ct values across the dataset so a reliable comparison is difficult, and the biological significance of this observation is unclear.
Other proximal residue replacements with potential relevance to Spike subunit function stability
Within the UK data and globally we have observed both independent instances of D614G and reversions back to 614D. It is unclear to what degree these variants are being transmitted and are circulating but the existence of reversions implies that the 614D is still relatively fit within individual hosts. We also observe two phylogenetic clusters of another replacement, 614N, within the UK suggesting independent origin transmission of this variant. The effect of 614N on Spike subunit function remains to be determined. Furthermore we observe replacements at the residues immediately adjacent to Spike 614 with Q613H co-occurring with and without D614G, V615I on the background of D614, and V615F co-occuring with D614G (Table 3). The replacements are associated with one or two UK clusters showing evidence for transmission within the UK. Spike 613H co-occurs with 614G and 614D representing possible convergent evolution. Spike 615I is largely constrained to Wales where it is associated with a large cluster but has not been observed since mid-April. Experimental studies will be required to determine whether these proximal mutations to site 614 have similar effects to 614G or, when co-occurring, are compensatory.
Circulating amino acid replacements found on Spike for amino acid residues 613-615. The ancestral haplotype is QDV. The table reports the number of distinct clusters that the respective genomes are found in.
Discussion
While Spike D614G has been shown to enhance the infectivity of pseudotyped lentiviruses carrying the SARS CoV spike protein in vitro [9,12], differences in cell infectivity will not necessarily be associated with greater within-host infectivity let alone transmissibility between hosts. The spread of a mutation in a population will be governed by epidemiological dynamics, patterns of global dispersion (founder effects), genetic drift/chance, as well as any potential fitness effects it confers if it enhances transmissibility. Available data do not allow precise measurement of the transmission fitness of the Spike 614G variant, but by using a large dataset of patient samples and a range of approaches we were able to detect some evidence for a difference in transmissibility. Not surprisingly given the many factors that contribute to transmission dynamics the values we have estimated are much less than the increase in cell infectivity measured in vitro.
Estimating epidemiological fitness of individual genetic variants during an emerging pandemic presents multiple challenges. Overall genetic diversity of SARS-CoV-2 is low, and most methods for identifying selection will have low sensitivity given the brief time over which evolution in human hosts has been observed. Significant positive selection has been detected at Spike 614 and other sites using statistical models based on the ratio of nonsynonymous to synonymous substitutions [19], however detection of positive selection does not necessarily imply any altered biology let alone enhanced transmissibility, and effects of individual mutations on transmissibility will generally be low [20]. Differences in transmission fitness can sometimes be indicated by convergent evolution resulting in homoplasies, but such approaches lack sensitivity for D614G as almost all circulating 614G lineages derive from a single ancestor[14]. Our finding of substitutions in neighbouring sites (Spike V615I etc.) and the appearance of D614N is, however, suggestive of a more complex selective landscape in this region of the Spike protein than was first indicated. We also note that our analysis is limited by necessity to comparison of co-circulating lineages with multiple substitutions shared by common descent (not only D614G), and it is impossible to disentangle effects of substitutions shared between lineages. One amino acid replacement is notable: RdRp P323L, occurred almost concurrently with D614G, and is in almost perfect linkage equilibrium with 614, although the biological effect and likely significance of this change is yet to be determined [21].
We have drawn on two sources of information regarding growth of the Spike 614G variant: The relative frequency of 614G and 614D over time and changes in genetic diversity of 614G and 614D lineages (phylodynamics). In exponentially growing populations, changing frequency of one variant can indicate difference in fitness, but the rate of 614G importations into the UK also increases over time, making direct comparisons challenging. We have controlled for this effect using phylogenetic analysis and only counting samples derived from cocirculating clusters representing distinct introductions into the UK. Separately, phylodynamic methods allow inference of growth and decline of effective population size, and we have compared growth patterns for spike 614G and 614D lineages. These phylodynamic estimates are imprecise, and we have not detected a consistent difference using phylodynamic methods. We have observed, however, that 614G clusters tend to grow to a larger size than 614D clusters after controlling for time of introduction into the UK. This is consistent with a transmission advantage of 614G variants, but could also be the result of unknown confounders which increase the probability that 614G lineages will be sampled. Our data will be naturally biased towards samples which are easy to sequence, and we have observed a very slight but significant downwards shift in Ct of 614G. This may explain the age association observed with lower-viral loads being harder to detect in younger people, while in older individuals age-associated host-properties dominate.
Increasing frequency of sampling 614G is consistent with a selective advantage of this variant, however several limitations of the data and analysis should be considered when interpreting this finding. We have applied classic population genetic models premised on contrasting exponential rates of growth of the 614G and 614D populations while controlling for founder effects, but in reality the SARS-CoV-2 epidemic is both noisier and highly structured in ways not accounted for in this model. The frequency of 614G and 614D variants can change rapidly due to stochastic fluctuations, especially early in the epidemic. The sampling process is also inhomogeneous through time and often reactive to short-term public health emergencies rather than surveillance. Most of the sequencing performed by centres in the UK is focused on symptomatic cases, often using diagnostic residual samples. As testing priorities change, and as cases in different segments of the population fluctuate, signals may emerge that are due to policy rather than biology.
Phylodynamic estimates of reproduction numbers are sensitive to the context of early spread of epidemic clusters which may have involved superspreading events [22]. These events are highly variable and phylodynamic methods are inherently imprecise with poorly resolved phylogenies. The Spike 614 polymorphism explains little variance in the rate of spread of individual clusters, but a pattern is discernable at the epidemic level. Incorporating additional information about the frequency of sampling 614G and 614D variants improved precision of phylodynamic estimates. Phylogenetic clusters are skewed towards successful colonization events which initiate large outbreaks. These analyses do not reflect cross-border transmission that failed to initiate large outbreaks and these failed introductions comprise a large proportion of clusters. Estimates of the reproduction number based on large clusters are not representative of the epidemic as a whole and may be larger on average.
Case fatality rates and infection fatality rates seem to vary widely from country to country and over time. It is unclear to what degree this reflects difficulties in collecting these statistics reliably or actual differences in underlying mortality due to factors such as the age structure of the population[23]. Reassuringly, and in agreement with a previous study [9], we do not detect a difference in virulence between Spike 614 variants. By estimating mortality rates as opposed to rates of hospitalization or ICU care our results complement those in [9] and are based on a substantially greater sample size. A small but significant association with age may indicate minor differences in clinical outcome or frequency of symptomatic infection which bears further study. The data is skewed towards hospitalized cases which represents the severe end of the disease spectrum and it is not possible to evaluate mild differences in virulence, particularly in younger individuals, which may be present in the larger population of mild and asymptomatic infections.
Our analysis emphasises that while laboratory experiments can identify changes in virus biology, examining the magnitude of phenotypic effects of mutations in vitro needs to be cautiously extrapolated to predictions about transmission at a population level. In the case of D614G, a large increase in cellular infectivity results in a weak population-level signal that nonetheless produces a discernible effect on transmissibility. While we believe an effect on SARS-CoV-2 transmissibility, caused by D614G, is likely to be present, it is important to note that the estimation of the absolute size of this effect is uncertain and much harder to predict. Although the signal is difficult to detect, the unprecedented size of our UK dataset, combined with comprehensive metadata, enables many potential biases within the data to be controlled for. This work is therefore demonstrative of the value of large-scale coordinated sequencing activities to understand a pandemic in real time.
This study and complementary work by others shows that the transmissibility of SARS-CoV-2 can change as a pandemic unfolds. Whether the current explosive epidemics currently being experienced across the world are to any degree being driven by D614G or it is simply the beneficiary of being in the right place at the right time, it is now the dominant variant. Changes in the transmissibility of a circulating virus could have a major effect on pandemic planning and the effectiveness of pandemic response, so it is critical that the model parameters in use for planning are based on the currently circulating virus. Work on vaccines, therapeutics and other interventions must allow for this but also keep in mind that reversions, and other mutations at the same or adjacent residues, will undoubtedly emerge in the future.
Methods
Sample collection and sequencing
We utilized data from the Coronavirus Disease 2019 (COVID-19) Genomics UK Consortium (CoG-UK)[16], a partnership of more than 18 academic, medical and public health research centres contributing sequencing and analysis capabilities. Sequence data was generated from a variety of protocols and platforms and were uploaded to a centralized environment for storage and analysis (MRC-CLIMB) (https://www.climb.ac.uk/)[24]. Data are uploaded with a standard set of clinical and demographic metadata and information about sequencing protocols and sample collection methods. Data undergo quality control and assembly and lineage assignment [25]. Data which complete quality control and assembly steps are released on a weekly basis. Sequence data are periodically shared through two open access databases, the European Bioinformatics Institute[26] and the Global Initiative on Sharing All Influenza Data[27]. We utilized 26,986 whole genome sequences contained in the June 19 release (https://www.cogconsortium.uk/data/) and for which the Spike 614 genotype could be determined and sample collection date was known.
Phylogenetics and calculation of maximum parsimony clusters
Maximum likelihood (ML) phylogenetic trees were estimated separately using IQTree v1.6.12 for major global lineages [25,28]. Phylogenies were rooted on a sample from the ancestral lineage. UK clusters were identified using parsimony-based ancestral state reconstruction [29] with internal nodes classified as UK or non-UK. Most UK clusters are descended from polytomies with descendents in multiple countries, and reconstruction of ancestral states at such nodes is ambiguous. In such cases the polytomy node was assigned the same state as it’s ancestor. We consider two extremes of the maximum parsimony method for reconstructing ancestral states at bifurcating nodes: Accelerated transition (AT) which favours transitions to the UK as close to the root of the tree as possible, and delayed transition (DT) which favours transition to the UK as far from the root as possible. Unless otherwise stated, results are based on DT clusters which are more likely to represent transmission within the UK.
Statistical analyses
Size of clusters was evaluated using log-linear multivariate regression. Effect of genotype on phylodynamic growth rates was estimated using multivariate weighted regression. Regression weights are inversely proportional to precision of estimated growth rates. Univariate comparisons used the Kruskal Wallis test. Kernel density estimation of sample time distributions used gaussian kernels and a bandwidth of 2 days. Statistical models were implemented in R 3.6.3.
Logistic growth model
According to this model the number of infected with the Spike 614D variant grows exponentially at a rate r and the number with the Spike 614G variant grows exponential at rate r(1 + s). If NX is the number infected initially with variant X, the proportion of the population with Spike 614G at time t is
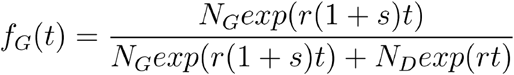
This model can be fitted to a sequence of sample times with(t1,…, tn) with Spike 614 genotypes (y1, …, yn) by maximum likelihood. The objective function is
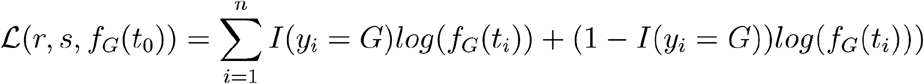
Formally, fitting this model is equivalent to logistic regression of genotype on time where the coefficient corresponds to the compound parameter ρ = r × s. Deriving the selection coefficient therefore requires additional information about the growth rate. We consider a range of plausible values for this rate corresponding to a reproduction number in the range 2.0-3.5 and a serial interval of 6.5 days[18]. The final confidence interval is based on these ranges as well as the confidence interval of ρ computed using profile likelihood.
Phylodynamic analysis of cluster growth rates with parametric coalescent model
We used a two-epoch coalescent model to estimate a period of exponential growth followed by an independently estimated period of exponential decline. Note that although we refer to growth and decline, the growth rates for both epochs can take either positive or negative values. The transition time from growth to decline was estimated independently for each cluster using a normal prior with a mean of the 23rd March 2020 (2020.2254), the date of ‘lockdown’ in the UK, and a standard deviation of two weeks. The data consisted of delayed transition clusters of more than 40 sequences as of the 19th June 2020.
A normal hyperprior is specified for cluster growth/decline rates for each genotype and the mean and precision of the hyperprior are estimated. The posterior mean growth/decline rates for each genotype are estimated along with the growth/decline rate for each cluster individually.
Posterior growth rates within each genotype are therefore correlated. The prior for the mean growth rate is Normal(0,100/year) and the prior of the precision parameter is Gamma(1,0.001).
We compute the selection coefficient from growth rates with the formula 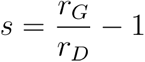 where r is the mean growth rate for each group of clusters.
where r is the mean growth rate for each group of clusters.
The model was implemented in BEAST v1.10.5[30]. Two independent chains of 100m states were run for each variant, with 10% removed from each chain to account for burn-in. Convergence was assessed using Tracer[31] prior to further analysis. The HKY model was used to model nucleotide evolution[32], and, following Duchene et al.[33], the evolutionary clock rate was fixed at 0.001 substitutions per site per year. Other priors used are described in table S1.
Phylodynamic analysis of cluster growth rates with non-parametric coalescent model
Rooted and dated phylogenies were estimated by randomly resolving polytomies in the ML trees described above using ape 5.3[34] and treedater 0.5.1[35]. The mean clock rate of evolution was constrained to (0.00075,0.0015). Branch lengths were smoothed by enforcing a minimum number of substitutions per site on each branch and by sampling from the distribution estimated by treedater. This was carried out 20 times for each UK lineage. Growth rates were estimated using skygrowth 0.3.1[36] using Markov chain Monte Carlo (MCMC) and 1 million iterations for each time tree and using an Exponential(10−4) prior for the smoothing parameter. The final results were produced by averaging across 20 time trees estimated for each cluster. Code to reproduce this analysis is available at https://git.io/JJkIM.
Model-based phylodynamic analysis
We applied a susceptible-exposed-infectious-recovered (SEIR) model[37] for the SARS-CoV-2 epidemic in London linked to an international reservoir. The SEIR model assumed a 6.5 day serial interval. The estimated parameters included the initial number infected, the susceptible population size, and the reproduction number. The model included bidirectional migration to the region outside of London (both within the UK and internationally) at a constant rate per lineage. Evolution outside of London was modelled using an exponential growth coalescent. Additional estimated parameters include the migration rate, and the size and rate parameters for the exponential growth coalescent. This model was implemented in the BEAST2 PhyDyn package[38,39] and is available here: https://git.io/JJUZv. The phylogenetic tree was co-estimated with epidemiological parameters. In order to make results comparable between 614D and 614G lineages, the molecular clock rate of evolution was fixed at a value estimated using all data in treedater 0.5.1. Nucleotide evolution was modeled as a strict clock HKY process [32]. To fit the model we ran 20 MCMC chains for 20 million iterations, each using 4 coupled MCMC chains [40]. Bespoke algorithms were used to exclude chains which failed to sample the target posterior. We used identical uninformative Lognormal(mean log=0, sd log = 1) priors for the reproduction number in 614G and 614D lineages.
The model was fitted to 614G and 614D sequence data separately before being combined for joint inference with the sample frequency data. This is carried out using a sampling-importance-resampling strategy[41]. We sampled parameters from the posterior estimated from genetic data uniformly and computed importance weights using a sequential Bernoulli likelihood based on the estimated frequency of 614G and 614D over time. Parameters resampled 1 million times with these weights yield our final estimate of the posterior.
The selection coefficient given a ratio of reproduction numbers is computed as follows:
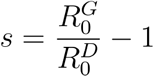
Data Availability
All sequence data and metadata used in this work is shared via: The COG-UK website : https://www.cogconsortium.uk/data/ GISAID: https://www.gisaid.org/ and the ENA as part of Bioproject PRJEB37886: https://www.ebi.ac.uk/ena/data/view/PRJEB37886
Acknowledgements
We thank all partners and contributors to the COG-UK consortium who are listed at https://www.cogconsortium.uk/about/. We also acknowledge the important work of SARS-CoV-2 genome data producers globally contributing sequence data to the GISAID database, and particularly acknowledge the groups who have generated data used by this project, listed in Table S3. EV acknowledges the MRC Centre for Global Infectious Disease Analysis MR/R015600/1. VH was supported by the Biotechnology and Biological Sciences Research Council (BBSRC) [grant number BB/M010996/1]. JTM, RMC, NJL and AR acknowledge the support of the Wellcome Trust (Collaborators Award 206298/Z/17/Z – ARTIC network). AR is supported by the European Research Council (grant agreement no. 725422 – ReservoirDOCS). DLR is supported by the MRC (MC_UU_1201412). J.S. was supported by the Biotechnology and Biological Sciences Research Council-funded South West Biosciences Doctoral Training Partnership [training grant reference BB/M009122/1]. TRC and NJL acknowledge support from the MRC which funded computational resources used by the project [grant reference MR/L015080/1]. TRC acknowledges funding as part of the BBSRC Institute Strategic Programme Microbes in the Food Chain BB/R012504/1 and its constituent projects BBS/E/F/000PR10348 and BBS/E/F/000PR10352]. AP and TRC acknowledge support from Supercomputing Wales, which is part-funded by the European Regional Development Fund (ERDF) via Welsh Government. The project was also supported by specific funding from Welsh Government which provided funds for the sequencing of a subset of the Welsh samples used in this study, via Genomics Partnership Wales.
Footnotes
References



