
Abstract
Coronavirus disease 2019 (COVID-19) was first diagnosed in Colombia from a traveler arriving from Italy on February 26, 2020. To date, available data on the origins and number or introductions of SARS-CoV-2 into the country are limited. Here, we sequenced SARS-CoV-2 from 43 clinical samples and—together with another 73 genomes sequences available from the country—we investigated the emergence and the routes of importation of COVID-19 into Colombia using epidemiological, historical air travel and phylogenetic observations. Our study provided evidence of multiple introductions, mostly from Europe, with at least 12 lineages being documented. Phylogenetic findings validated the linkage of epidemiologically-linked transmission chains. Our results demonstrate the advantages of genome sequencing to complement COVID-19 outbreak investigation and underscores that human mobility and genetic data are relevant to complete epidemiological investigation and better characterize virus transmission dynamics at local scales.
Introduction
Coronavirus disease 2019 (COVID-19) is a life-threatening respiratory illness caused by SARS-CoV-2, an emerging zoonotic virus first identified in Wuhan city, Hubei province, China (1). The first confirmed cases of COVID-19 were reported on January 12, 2020, from patients presenting respiratory symptoms between December 8, 2019 and January 2, 2020 (2). Despite early containment and mitigation measures set in place (3), the high infectiousness, presymptomatic and prolonged transmission of SARS-CoV-2 (4,5) combined with other factors such as globalization, led to its rapid spread across the world.
Rigorous contact-tracing and physical distancing measures implemented in different countries have been demonstrated to be effective in delaying the epidemic during the contention phase (6–9). However, ensuing lockdowns and travel restrictions to minimize the healthcare burden has led to wellbeing decline and economic downturn, with profound impacts in low-to-middle income countries (10). The contention phase in Colombia started with the first case of COVID-19 confirmed by the Instituto Nacional de Salud (INS) on March 6, 2020, from a person returning from Italy on February 26, 2020 (11).On March 23, 2020, 314 cases had been confirmed which prompted the closure of all the country borders to contain the outbreak. On March 31, 2020, more than 10% of confirmed cases were missing from the contact-tracing (infections linked to unknown sources), and therefore the mitigation phase was declared with physical-distancing as the main strategy to further limit virus spread. On June 18, 2020, 57,046 confirmed cases, and 1,864 deaths had been reported in Colombia (12).
The unprecedented global health and societal emergency posed by the COVID-19 pandemic urged unprecedented data sharing and faster-than-ever outbreak research developments which are reflected in the fact that over 37.000 SARS-CoV-2 complete genomes have been made available through public databases, mainly GISAID (Global Initiative on Sharing All Influenza Data). Genome availability and the fact that SARS-CoV-2 is an RNA virus with an estimated substitution rate of around 0.8×10−3 to 1.1×10−3 substitutions/site/year (13,14), enabled us to detect the virus is fast generating genetic variation as it is transmitted, and therefore genomic epidemiology is a powerful approach to characterize the outbreak (15). This approach which relies on phylogenetic analysis has allowed researchers across the world to ascertain SARS-CoV-2 emergence in humans, to track its geographical spread and to investigate its evolutionary dynamics (16–20).
Here we describe the complete genome sequencing of SARS-CoV-2 from 43 clinical samples together with the epidemiological investigation of imported cases and the phylogenetic findings using 122 genome sequences from Colombia, which characterize the epidemic onset of COVID-19 in the country. Our study suggests multiple independent introductions from across the world, mainly from Europe and the co-circulation of multiple lineages in the most affected departments. These findings contribute to the molecular surveillance of COVID-19 and provide venues for future outbreak response and interventions in the country.
Materials and methods
Ethical statement
The INS is the reference lab and health authority of the national network of laboratories. In case of a public health emergency or when scientific research in public health is required, the national law 9/1979, decrees 786/1990 and 2323/2006, authorizes the INS to use the biological material and associated epidemiological information without informed consent, including the anonymous disclosure of results. This study was performed following the ethical standards noted in the 1964 Declaration of Helsinki and its later amendments. The information used for this study comes from secondary sources of data that were anonymized and do not represent a risk to the community.
Sample preparation
Nasopharyngeal swabs samples from patients with clinical presentations of SARS-CoV-2 across the country were received at the Instituto Nacional de Salud (INS, National Institute of Health) as part of the virological surveillance of COVID-19. A total of 43 samples were selected for genome sequencing representing either the earliest documented cases, findings from outbreak investigation (linked to transmission chains), and country-wide representativeness. Viral RNA extraction was performed with the QIAamp Viral RNA Mini kit (Qiagen Inc., Chatsworth, CA, USA) using the MagNA Pure LC nucleic acid extraction system (Roche Diagnostics GmbH, Mannheim, Germany).
Genomic library preparation and sequencing
Library preparation and sequencing were performed following the ARTIC network (real-time molecular epidemiology for outbreak response) protocol and both Nanopore and Illumina technologies (21). In brief, cDNA was synthesized with the SuperScript IV reverse transcriptase (Thermo Fisher Scientific, Waltham, MA, USA) and random hexamers (Thermo Fisher Scientific, Waltham, MA, USA). A set of 400-bp tiling amplicons across the whole genome of SARS-CoV-2 were amplified with the primer schemes nCoV-2019/V3 (22).
Amplicons were pooled and quantified with the Qubit dsDNA High Sensitivity assay using the Qubit 3.0 instrument (Life Technologies, Carlsbad, CA, USA) and then purified with AMPure XP beads (Beckman Coulter, Brea, CA, USA). Ten amplicons were processed using Oxford Nanopore Technologies (Oxford Nanopore Technologies, Oxford, UK). These samples were barcoded with the Native Barcoding Kit EXPNBD104 (Oxford Nanopore Technologies, Oxford, UK) and pooled in an equimolar amount. Genomic libraries were prepared with the ligation kit 1D SQK-LSK109 (Oxford Nanopore Technologies, Oxford, UK) and sequenced using a FLO-MIN106-R9.4 flow cell and the MinION instrument (Oxford Nanopore Technologies, Oxford, UK). The remaining 33 amplicons were processed using illumina technologies. Genomic libraries were prepared with the Nextera XT DNA library prep kit (Illumina, San Diego, CA, USA) and sequenced using the MiSeq reagent kit version 2 and the MiSeq instrument (Illumina, San Diego, CA, USA).
Genomic sequence assembly
Nanopore reads were basecalled using Guppy version 3.2.2 (Oxford Nanopore Technologies, Oxford, UK) and then demultiplexed and trimmed using Porechop version 0.3.2_pre (23). Processed reads were aligned against SARS-CoV-2 reference genome (GenBank NC_045512.2) using BWA-MEM (24). Single nucleotide variants were called with a coverage depth of at least 20x and then polished consensus was generated using Nanopolish version 0.13.2 (25). MiSeq reads were demultiplexed and quality control was performed with a Q-score threshold of 30 using fastp (26). Processed reads were aligned against SARS-CoV-2 reference genome (GenBank NC_045512.2) and consensus genomes were generated using BWA-MEM version 0.7.17 (24) and BBMap (27).
Phylogenetic analysis of SARS-CoV-2 in Colombia
The SARS-CoV-2 genome sequences obtained in the present study and those from Colombia deposited in GISAID were collated (n=122) and combined with 1461 representative genome sequences from the South America-focused subsampling available from NextStrain (28) as in 20-May-2020 (Suppl Table 1) plus reference MN908947.3. The full genomic dataset was classified in lineages (29) using PANGOLIN (Phylogenetic Assignment of Named Global Outbreak LINeages) (30) and aligned with 10 iterative refinements using MAFFT (31). All the alignment positions flagged as problematic for phylogenetic inference were removed (e.g. highly homoplasic positions and 3’ and 5’ ends) (32). Maximum likelihood phylogenetic reconstruction was performed with the curated alignment and a HKY+G4 substitution model (33) using IQTREE (34). Branch support was estimated with an SH-like approximate likelihood ratio test (SH-aLRT) (35). Six sequences from Colombia that showed inconsistent temporal signal with a clock analysis using TreeTime (36) were removed from further analysis. Time-scaled trees were inferred and rooted with least-squares criteria and the evolutionary rate of about 1.1×10−3 subs/site/year estimated by (13) using TreeTime (36) and LSD (37). The geographical locations (aggregated by continent except for Colombia) of the sequence data were considered as discrete states and used for migration inference—with transitions modeled as a time reversible process— in TreeTime (36). The number of state transitions into Colombia were interpreted as a proxy for the minimum number of introductions. In order to measure the effect of the SARS-CoV-2 uneven genomic representativeness across the world, two downsampling strategies datasets were implemented where, based on location, the sequences were randomly resampled 100 times and the phylogenetic and migration inference was replicated. The downsampling strategies were as follows: (i) retaining a number of sequences per region (whenever possible) equal to the number of sequences available for Colombia and (ii) retaining 50 sequences per region and the total number of sequences from Colombia (this was the most even sampling per region for the South America-focused subsample).
Potential routes of COVID-19 importation in Colombia
The relative proportion of expected importations of COVID-19 by country to Colombia was inferred by taking into account SARS-CoV-2 incidence per international air passengers arriving in Colombia and the available flight travel. The number of international flights and number of passengers arriving to Colombia from January 1 to March 9, 2020 were obtained from the Special Administrative Unit of Civil Aeronautics of Colombia (Aerocivil, aerocivil.gov.co). The air travel data consists of direct flights from 14 countries to 7 main cities. COVID-19 incidence for each of the 14 countries with direct flights to Colombia was calculated using the number of confirmed cases reported by the World Health Organization—as of March 17, 2020, the date when travel restrictions started in Colombia (38)—and the total population for each country for 2019 reported in the United Nations World Population Prospects 2019 database (39) as in (40) (Appendix).
Results and discussion
Epidemiology investigation of introductions, contact-tracing and community transmission of SARS-CoV-2 in Colombia
In Colombia, preventive isolation and monitoring for passengers arriving from China, Italy, France, and Spain started on March 10, 2020. National health emergency was declared on March 12 and tougher measures started to be set in place including the closing of borders (March 17) the ban of international (March 20) and domestic flights (March 25). Implementations of lockdowns occurred from March 25 onwards (Resolutions 380 and 385 from the Colombian Ministry of Health and Social Protection; Decree 412 and 457 from the Colombian Ministry of the Interior; Decree 439 from the Ministry of Transport). Despite massive drop in air traffic, more than 15.500 residents returned to Colombia through humanitarian flights during April and June (41).
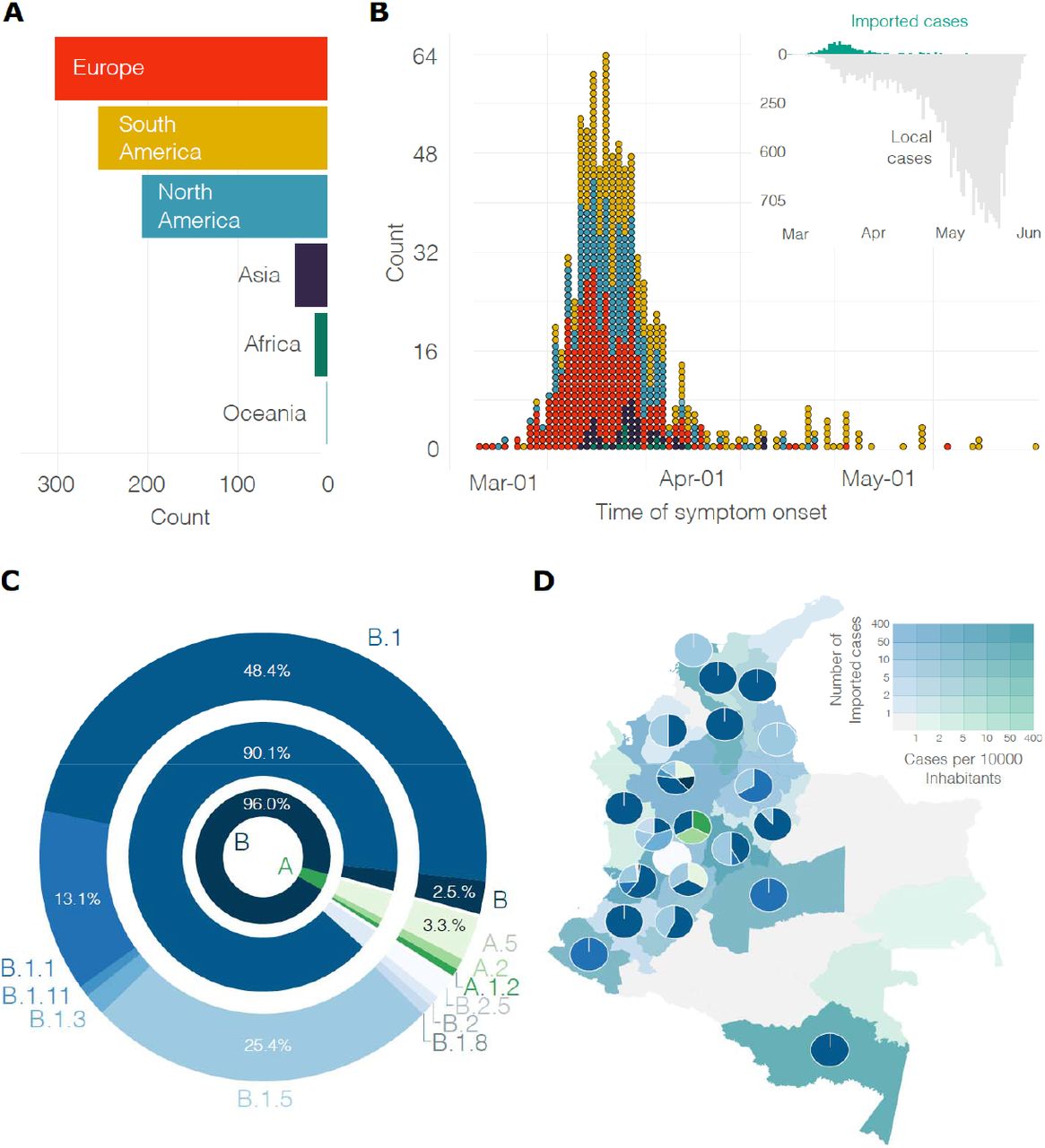
As per June the first, over 30 thousand cases of COVID-19 had been documented in the country and 857 cases (2.8%) had been linked abroad (Figure 1A). Forty-eight (4.8%) of the imported cases were asymptomatic and detected through contact-tracing; the majority of these cases were imported from Spain (39%) the United States (31.7%), Brazil (7.3%) and Mexico (4.9%). Most of the symptomatic imported cases were traced back to European countries and then to countries in the Americas. The most important geographical sources in terms of imported cases were Spain (30.5%), the United States of America (25.2%), Mexico (6%), Ecuador (5.8%), and Brazil (5.1%). The number of symptomatic imported cases steadily increased and reached a peak on March 14, 2020, when local cases were on the rise and earlier than the closure of borders and ban of international air travel. Our estimate is based on the average incubation time of COVID-19 (42) and consequently 4.8 days earlier than peak based on symptoms onset (March 18, Figure 1B). The first introductions were predominantly linked to Europe, however, both Europe and the American region were important geographical sources of infections during the onset of the epidemic. The introductions post-peak occurred for the most part from South American countries.

A) Region of origin for the reported imported cases. B) Distribution over time of the symptomatic imported and local cases colored by region (as in A). C) Frequency of the lineages (colores as in C) and sub-lineages of SARS-CoV-2 identified in Colombia. D) Distribution of lineages across the country; departments are colored by the number of imported cases per 10 thousand inhabitants and the number of reported introductions.
SARS-CoV-2 diversity in Colombia
In order to gain a better understanding of the dynamics of SARS-CoV-2 spread into Colombia, 43 whole genome sequences obtained in the present study were combined with sequences from Colombia deposited in GISAID, resulting in a set of 122 complete genomes. Sequences from Colombia were classified in a total of 12 sublineages: A.1.2, A.2, A.5, B, B.1, B.1.1, B.1.3, B.1.5, B.1.8, B.1.11, B.2, and B.2.5. Recent evidence suggests a role of the amino acid change D614G at the spike protein in conferring higher transmissibility (43). and the proportion of lineages documented in Colombia are likely due to founder effects and/or sampling strategy. For example, sublineages B.1, B.1.1, and B.1.5 were found in six of the early epidemiologically-linked transmission chains. Consistently, the most frequently observed lineages in Colombia were B.1 (n= 59, 48.4%), B.1.5 (n=31, 25.4%) and B.1.1 (n=16, 13.1%) (Figure 1C). Comparable findings were observed for the other South American countries (44–46) from the South America-focused subsampling available from NextStrain, where the most frequently observed lineages were B.1 (n= 149, 60.8%), B.1.5 (n=33, 13.5%) and A.5 (n=14, 5.7%). The median value of sequences available per department was 1.5 (mean of 3.9 sequences, a max of 45 sequences were obtained from a single locality). Consequently, on average, one single lineage was identified by department. For instance, the number of documented lineages was highly correlated with the availability of samples (Pearson’s product-moment correlation of 0.72, p-value<0.001) and uncorrelated with the number of local cases (Pearson’s product-moment correlation of 0.35, p-value=0.049). Consequently, five different lineages were documented in the departments of Valle del Cauca and Antioquia and three different lineages were documented in Cundinamarca which are the departments with a higher number of samples (Figure 1D) and whose capitals are the most populated. A moderate positive correlation was observed between the number of lineages documented in a department and the number of imported cases (Pearson’s product-moment correlation 0.51, p-value = 0.002).
Molecular evolution of SARS-CoV-2 in Colombia
A total of 133 nucleotide variants (NVs) were identified using the full genome sequences from Colombia and the reference NC_045512.2. The majority (n=131) of the NVs fell in the coding region (one NV was identified at each non-coding end). A total of 71 (54,2%) of the NVs in coding sites led to non-synonymous substitutions. Most of the NVs (92 out of 133) were unique to a sequence and among the shared NVs, 38 (out of 41) were associated with a specific lineage (Tables S2 and S3). These observations suggest that the substitutions are not laboratory-specific and most likely the outcome of in-situ evolution and/or shared ancestry. (Appendix).
Evolutionary relationships between local and global SARS-CoV-2 isolates
The time-stamped phylogeny of 122 isolates of Colombia and 1462 representative global SARS-CoV-2 isolates (Figure 2A) showed that the estimated time to the most recent common ancestor for the sampled sequence data is December 7, 2019 (October 25 - December 26). Asia was the inferred ancestral state at the root. Both these observations are in line with the known epidemiology of the pandemic. A root-to-tip regression of genetic distance against sampling time evidenced consistent temporal signal in the sequence data (Figure 2B). The isolates from Colombia appeared intersperse among the isolates from other countries (Figure 2A and 2C) suggesting multiple introductions. However, there was considerable phylogenetic uncertainty along the tree and the fine-grained relationships of the isolates from Colombia could not be resolved with confidence (Figure S1). Phylogenetic uncertainty together with uneven sampling made the quantification of the number of introductions into the country challenging, let alone dating the time of the introductions. The number of state transitions into Colombia (Figure 3A) heavily relies on the number and nature of the sequences that are included from other locations. Using all sequences, we estimated that on average 64 (IQ 62-67) introductions have occurred into the country but this estimate gets lower as we reduce the number of samples from other locations (down to 22 with the most even down-sampled dataset). Independently of the dataset and in line with the epidemiological information, most of the geographical source attributions of the introductions are attributed to Europe (Figure 3B and Figure S2). This observation is also in agreement with our estimates using travel data as detailed below (Figure 3C and Figure S2).

A) Time-resolved maximum likelihood tree of 1578 SARS-CoV-2 sequences including 122 from Colombia (red). B) Root-to-tip distance regression for the sequence data in A. C) Time-resolved maximum likelihood annotated by region of isolation. The outer ring represents SARS-CoV-2 lineages. The inner red ring highlights the relative position of the sequences from Colombia. The middle ring and the corresponding numbers indicate sequences from epidemiologically-linked transmission chains. Branches are colored by the geographical attribution from the migration inference. Highly supported groups are delineated by thicker solid lines.

A) The number of transition changes into Colombia following migration inference using all available sequences per region, retaining a number of sequences per region (whenever possible) equal to the number of sequences available for Colombia and 50 sequences per region and all sequences from Colombia. B) Geographical source attribution for every transition into Colombia derived from the migration inference using all the available sequences per region. C) Geographical contribution inferred using Air travel data per country aggregated by region.
Between January and March 2020, 7 cities of Colombia received 1,593,211 international passengers from 14 countries. Bogotá concentrated the majority of the flights with around 77% of the passengers, followed by Medellín with 11%, Cartagena with 6%, and Cali with 4%. In total, 35% of international passengers started their journey in the USA, 17% in Mexico, and 12% in Chile. However, we estimate that 87% of all imported COVID-19 cases came from Europe, 9.5% from North America, and 3.4% from South America. When stratified by country, the primary source of importation was Spain with 71.4% (Figure S2).of the cases, followed by the USA with 8.4%, Germany with 8%, and France with 3.4% 65.2% of the COVID-19 cases were expected to arrive at Bogotá, followed by Medellín with 20% and Cali with 9%. We estimate that the route Spain-Bogotá carried 42% of the total imported cases.
Since the first introduction of SARS-CoV-2 in Colombia (February 26, 2020) contact-tracing efforts had been set in place. Multiple sequences were obtained from seven distinct early epidemiologically-linked transmission chains. This information was mapped into the phylogeny (Figure 2C) and all but one set of sequences did not group. However, it appeared very close in the tree. This underscores the potential utility of genomic epidemiology to link individuals with incomplete information (e.g. cases that end-up disconnected due to intermediate asymptomatic carriers) and complement outbreak transmission.
This study has some limitations. Firstly, the geographical sources of infection relied on people to self-report their own symptoms onset and travel history, which is subject to inaccuracies. Secondly, we use air travel data from important destinations in Colombia but other locations may also have fueled COVID-19 emergence and dissemination in the country; flight travel data was not available from March 9, 2020, onwards. Thirdly, the number of sequences sampled represented a tiny fraction of the documented number of imported cases into Colombia; the sample was selected to be a country-wide representative given limited resources for genome sequencing and consequently, the introduced viral diversity may also have been underestimated. Another limitation of this analysis is the inherent uncertainty stemming from global unsystematic sampling. Therefore, the inferences about the number of introductions and the corresponding geographical source need to be interpreted with caution. We attempted to overcome this by undertaking sensitivity analyses and contrasting the results with the available epidemiological data and our estimates from travel data. However, more sequence data from Colombia and undersampled countries together with information of sampling representativeness per country is needed in order to account for sampling uncertainty in a more statistically rigorous manner.
Our study provided evidence that an important number of independent introductions occurred to Colombia with at least 12 lineages documented. Most of the notified introductions occurred from Europe and this observation was supported by phylogenetic and air travel data. Although the sequence data do not represent the actual number of epidemiologically-linked transmission chains, our phylogenetic findings validated the linkage for those epidemiologically-linked transmission chains with available sequence data. Our results further underscore the advantages of genome sequencing to complement COVID-19 outbreak investigation and support the need for a more comprehensive country-wide study of the epidemiology and spread of SARS-CoV-2 in Colombia.
Data Availability
Data supporting this paper can be accessed through GISAID at https://www.gisaid.org/ here and used under a Creative Commons Attribution (CC-BY) licence
Funding
This work was funded by Instituto Nacional de Salud. C.J.V-A is supported by an ERC Starting Grant (award number 757688). The funders had no role in study design, data collection, and analysis, decision to publish, or preparation of the manuscript.
Author Bio
Katherine Laiton-Donato is the head of the Sequencing and Genomics Unit, Dirección de Investigación en Salud Pública, Instituto Nacional de Salud, Colombia. Her research is focused on molecular virology of emerging viruses with impact in public health.
Figure captions
Table S1. GISAID’s nCoV-19 Acknowledgements.
Table S2. Nucleotide substitution patterns of the different lineages of SARS-CoV-2 circulating in Colombia.
Table S3. Amino acid change patterns of the different lineages of SARS-CoV-2 circulating in Colombia.
Table S4. Estimates of evolutionary divergence of SARS-CoV-2 over sequence pairs within and between lineages/sublineages from Colombia.
Figure S1. Schematic of the maximum likelihood tree of 1578 SARS-CoV-2 sequences. Branches with branch supports (approximate likelihood ratio test [aLRT]) below 0.7 were collapsed. Sequences from Colombia are indicated in red. Sequences from other regions in polytomies were reduced to a maximum of three sequences and represented as fading taxa.
Figure S2. Potential countries of introductions of SARS-CoV-2. A) Geographical source attribution for every transition into Colombia derived from the migration inference retaining a number of sequences per region (whenever possible) equal to the number of sequences available for Colombia and retaining 50 sequences per region and all sequences from Colombia. B) Number of passengers arriving in Colombia from different countries. C) Geographical contribution inferred using Air travel data per country.
Acknowledgments
The authors thank the National Laboratory Network and Virology Group of INS for routine virologic surveillance of SARS-CoV-2 in Colombia. The authors thank all researchers who deposited genomes in GISAID’s EpiCoV (TM) Database contributing to genomic diversity and phylogenetic relationship of SARS-CoV-2. The authors thank Maylin Gonzalez Herrera for her technical assistance.




{kind=link}
{kind=link}
{kind=link}