
Abstract
The ongoing severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic has caused a public health crisis that is exacerbated by our poor understanding of correlates of immunity. SARS-CoV-2 infection can cause Coronavirus Disease 2019 (COVID-19), with a spectrum of symptoms ranging from asymptomatic carriage to life threatening pneumonia and cytokine dysregulation [1–3]. Although antibodies have been shown in a variety of in vitro assays to promote coronavirus infections through mechanisms requiring interactions between IgG antibodies and Fc gamma receptors (FcγRs), the relevance of these observations to coronavirus infections in humans is not known [4–7]. In light of ongoing clinical trials examining convalescent serum therapy for COVID-19 patients and expedited SARS-CoV-2 vaccine testing in humans, it is essential to clarify the role of antibodies in the pathogenesis of COVID-19. Here we show that adults with PCR-diagnosed COVID-19 produce IgG antibodies with a specific Fc domain repertoire that is characterized by reduced fucosylation, a modification that enhances interactions with the activating FcγR, FcγRIIIa. Fc fucosylation was reduced when compared with SARS-CoV-2-seropositive children and relative to adults with symptomatic influenza virus infections. These results demonstrate an antibody correlate of symptomatic SARS-CoV-2 infections in adults and have implications for novel therapeutic strategies targeting FcγRIIIa pathways.
Antibody responses to viral infections in humans are varied and of widely divergent clinical significance. Antibodies formed early during infection can bind to virus particles, forming immune complexes that may neutralize or mediate clearance of virus. On the other hand, immune complexes can also promote inflammation and exacerbate symptoms of disease via interactions between antibody Fc domains and FcγRs. How antibodies within immune complexes modulate infection depends, in part, on their Fc domain structure. Fc structure, in turn, dictates interactions with Fc receptors that are expressed by a variety of cells that become activated during infection.
Antibody isotypes, IgG, IgA, and IgM are a primary determinant of structure and thus of activity. IgM production occurs first and signals the onset of new B cell responses. Production of class-switched antibodies follows, with IgA central in mucosal immunity and IgG being the dominant isotype in systemic antiviral immunity. IgG effector function is governed by Fc-FcγR interactions which are regulated by IgG subclasses (IgG1, IgG2, IgG3, IgG4) and post-translational modifications of antibodies within immune complexes. Importantly, individuals produce distinct structural repertoires of IgG Fc domains, with some people producing highly activating repertoires enriched for IgG1 and IgG3 and/or reduced core-fucosylation of the IgG1 Fc domain. In most infections, IgG responses are either protective in nature or do not have a significant impact on infection [8]. However, in rare circumstances antibodies with specific Fc structures may cause cell activation or pro-inflammatory activity during infections that exacerbate symptoms of disease. A striking example of this is dengue virus infections that occur in the presence of reactive, non-neutralizing IgGs. Those with severe dengue disease disproportionately produce antibodies with reduced fucosylation of the Fc; this modification enhances affinity of the Fc for the activating FcγR, FcγRIIIa which modulates dengue disease pathogenesis, likely through multiple pathways [9, 10].
It remains unknown whether there are any specific features of antibodies produced by patients with COVID-19 disease that might enhance or mitigate disease symptoms. To address this, we performed a global analysis of antibodies produced during SARS-CoV-2 infections. Children are rarely diagnosed with COVID-19 disease, and almost never develop severe COVID-19 illness despite being susceptible to productive infections [11–14]. Thus, we reasoned that it would be informative to compare antibodies produced by adults with symptomatic, PCR-diagnosed COVID-19 and antibodies produced by children who were seropositive for SARS-CoV-2 infections but had no COVID-19 diagnosis. To this end, we performed an enzyme-linked immunosorbent assay (ELISA)-based screen on remainder sera from the regional laboratory of a large Northern Californian health care system. Sera obtained between March 30th, 2020 to April 19th, 2020 from adult COVID-19 patients (n=225) and from children (n=802) without a COVID-19 diagnosis were assayed.
Sera were screened by ELISA for antibodies against the receptor binding domain (RBD) of the SARS-CoV-2 spike protein (Figure S1); all positive samples from children were validated in a secondary screen against the full-length spike protein, as previously described [15]. Sera from children and COVID-19 patients were considered positive if they reached a threshold of the average value of 130 historical negative controls plus four standard deviations. Overall, 1.87% of pediatric samples were positive for RBD and full-length spike antibodies whereas 74.2% of sera from PCR+ COVID-19 patients were seropositive for any RBD and full-length spike SARS-CoV-2 antibodies. Control sera from 12 subjects with documented seasonal coronavirus infections collected in early 2019 were negative for RBD-reactive antibodies (Figure 1A,B and Table 1).
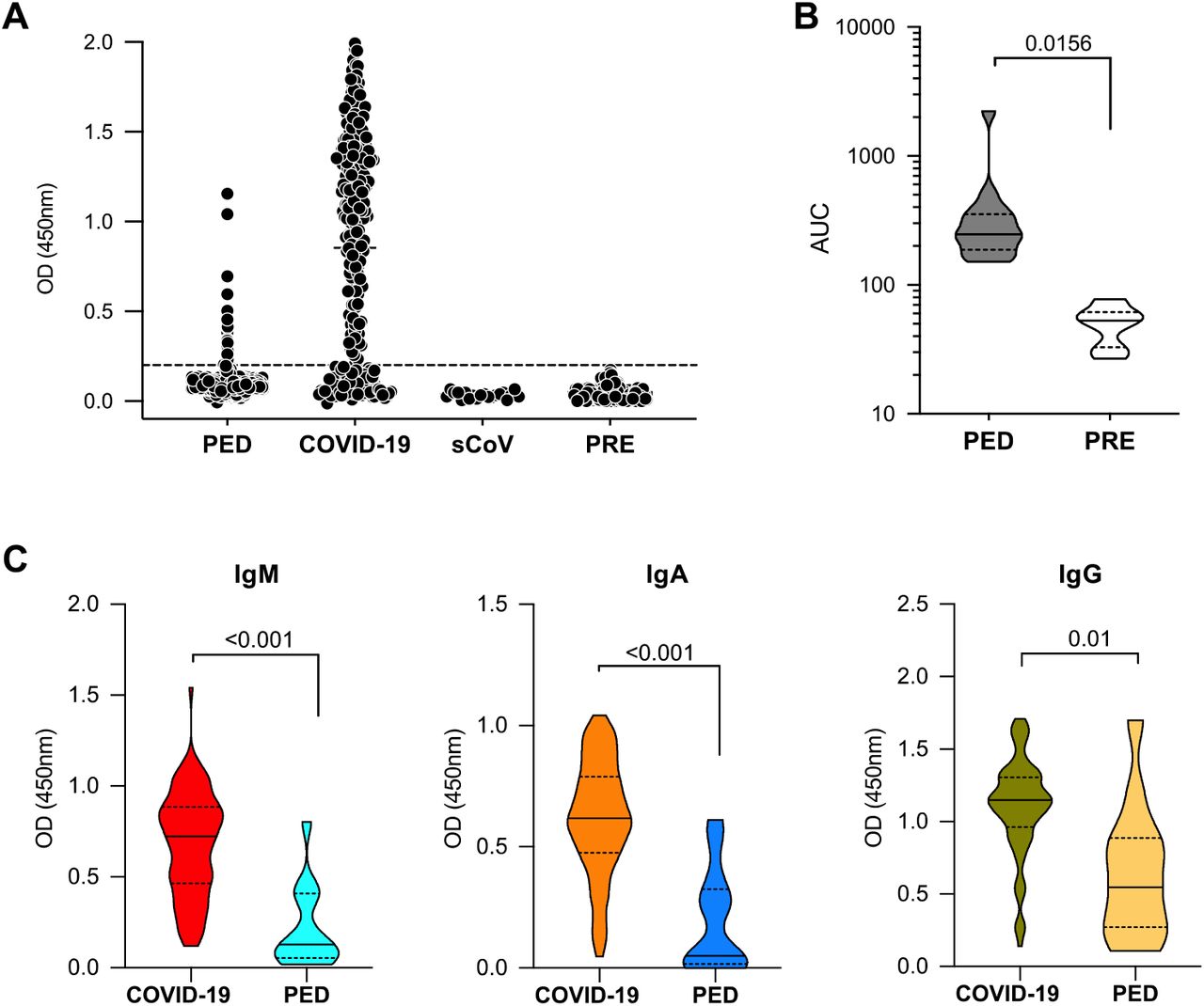
(A) Sera obtained between March 30th, 2020 and April 19th, 2020 from adult COVID-19 patients (COVID-19, n=225) and from children (PED, n=802) were assayed by ELISA for antibodies against the SARS-CoV-2 receptor binding domain (RBD). Control sera from 12 subjects with known seasonal coronavirus infections (sCoV) collected in early 2019 were included in the screen, along with 130 historical control samples derived from cohorts in the United States, Uganda and Nepal (PRE). (B) Anti-RBD positive sera were validated in a second screen that measures binding to the full spike protein from SARS-CoV-2. (C) RBD antibodies from seropositive COVID-19 patients and from seropositive children were characterized for isotypes. Known COVID-19 infections were associated with higher RBD antibody titers when compared with titers in the pediatric (PED) cohort. Data shown are representative of at least two experiments performed in duplicate or triplicate. P values were calculated using Wilcoxon rank sum tests. Violin plots show the distribution of sample values along with median (solid line) and quartile (broken line) values.
Samples studied were from adults with known COVID-19 Infections (PCR+ by nasopharyngeal swab) or from children (<18 years old) who were not diagnosed with COVID-19. Sera were collected at multiple hospltals/cllnlcs In Northern California during the SARS-CoV-2 pandemic.
With the SARS-CoV-2 seropositive subjects identified, we next characterized the isotype distribution of the RBD antibodies. Among our study subjects, COVID-19 patients had significantly higher serum titers of IgM, IgA and IgG anti-RBD antibodies compared with seropositive children (Figure 1C).
Next, we defined the determinants of anti-RBD IgG effector function in the COVID-19 patients and seropositive children. Anti-RBD IgG1, the most abundant IgG subclass, was characterized for post-translational modifications of the Fc using mass spectrometric methods [9, 10, 16]. Anti-RBD IgG1 from COVID-19 patients were significantly reduced in core fucosylation, galactosylation and bisection when compared with total IgG1 from 18 healthy adult controls. Anti-RBD IgG1 from COVID-19 patients also had significantly less Fc fucosylation relative to RBD IgG1 from children (Figure 2A). Reduced fucosylation was the only glycan modification that distinguished antibodies from COVID-19 patients and seropositive children. In addition to Fc glycosylation, IgG subclasses play a critical role in determining antibody effector function. We therefore quantified the relative abundance of IgG subclasses by mass spectrometry and found no difference between the subclass distribution of anti-RBD IgGs from COVID-19 patients and seropositive children compared with total IgG from healthy adults (Figure S2). This characterization revealed that anti-RBD IgG1 from COVID-19 patients was significantly different in Fc glycosylation relative to IgGs from healthy adults and was significantly reduced in fucosylation, specifically, relative to anti-RBD IgG1 from children.
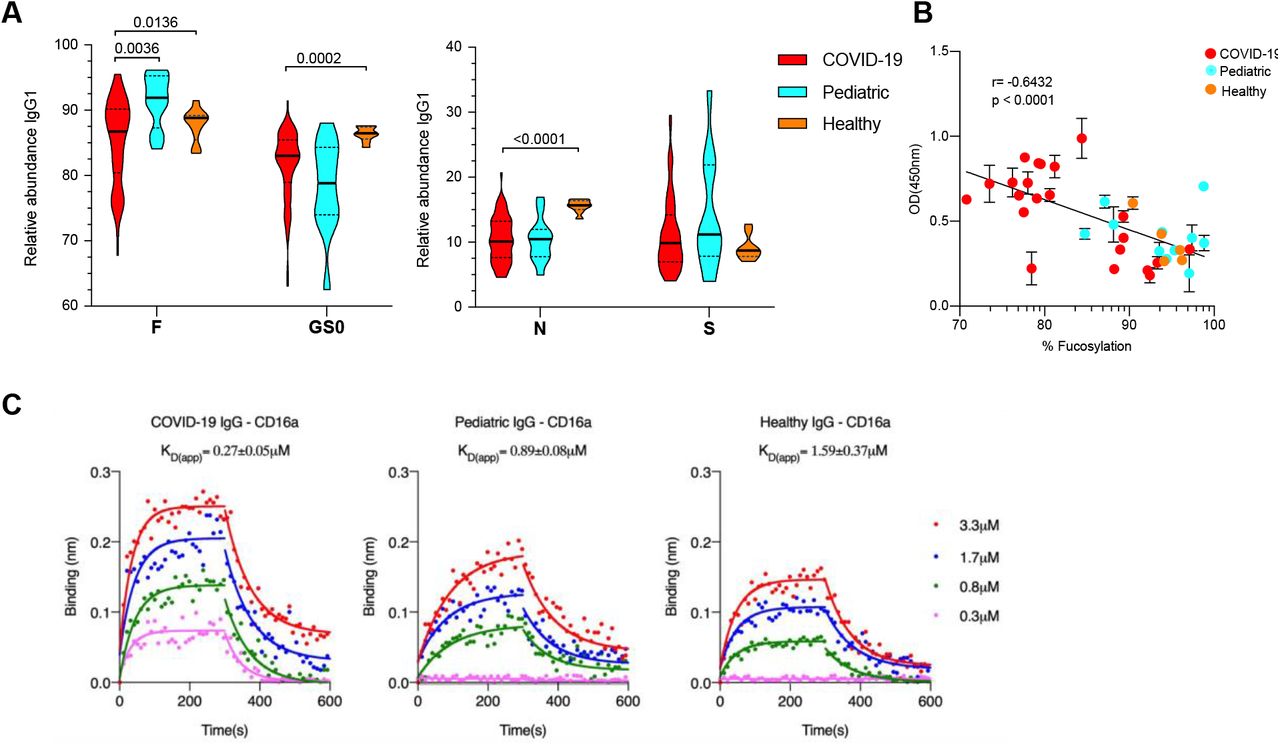
(A) Anti-RBD IgG1 were characterized for post-translational modifications of the Fc. IgGs from COVID-19 patients (red) were significantly reduced in fucosylation (F), galactosylation (GS0) and bisecting N-acetylglucosamine (N) when compared with total IgGs from healthy adult controls (orange). Anti-RBD IgGs from COVID-19 patients also had significantly less Fc fucosylation relative to RBD IgGs from children (light blue). No significant differences were observed in levels of Fc sialylation (S). (B) Correlation between the level of anti-RBD IgG1 Fc fucosylation and binding to FcγRIIIa. The binding of serum antibodies (n=38) to soluble FcγRIIIa correlated negatively with the degree of fucosylation (Pearson correlation coefficent r= –0.6432). Samples were representative of the spread of fucosylation over the dataset. (C) The apparent dissociation constants (KD(app)) for pooled IgGs purified from COVID-19 patients with anti-RBD IgG1 core fucosylation <80%, seropositive pediatric and historical healthy controls to FcγRIIIa/CD16a was determined by biolayer interferometry. The overlays of binding traces at different concentrations (3.3uM followed by 2-fold dilutions) of the analytes are shown (solid circles). The fits are indicated by solid lines. The assay was replicated in two independent experiments and shown are representative traces from one experiment. P values in (A) were calculated using Wilcoxon rank sum tests. Violin plots in (A) show the distribution of sample values along with median (solid lines) and quartile (broken lines) values.
Reduced IgG1 Fc fucosylation confers 5–10-fold higher affinity for FcγRIIIa, on a monomeric basis, between IgG and FcγRIIIa [17]. This receptor is present on monocytes, macrophages and NK cells and can enhance cell activation, pro-inflammatory cytokine production and cytotoxic effector cell activity [18, 19]. To determine whether serum IgGs from the study subjects differed in their capacity to bind FcγRIIIa, we performed an ELISA to measure binding by serum IgG to recombinantly expressed FcγRIIIa (F158). 38 sera were selected that represented the range of fucosylation over the sample set. We observed that the amount of anti-RBD IgG1 fucosylation in serum negatively correlated with binding to FcγRIIIa (Figure 2B). Next, to better characterize differences in IgG-FcγR interactions associated with differences in fucosylation, we determined the apparent dissociation constant of purified serum IgGs to soluble FcγRIIIa (sFcγRIIIa). IgGs were pooled from COVID-19 patients with anti-RBD IgG1 fucosylation < 80%, from seropositive pediatric subjects and from historical healthy controls (n=7 sample per pool). Consistent with previous results [20], we observed that IgGs with lower core fucosylation bound sFcγRIIIa with higher affinity (3–6 fold) (Figure 2C).
We next asked whether levels of Fc fucosylation in COVID-19 patients were comparable to those observed in another respiratory viral infection caused by an influenza A virus. To do this, we compared anti-hemagglutinin (HA) and anti-neuraminidase (NA) IgG1 from 29 symptomatic influenza virus patients who underwent controlled infection with the influenza virus A/California/04/2009/H1N1 [21]. COVID-19 patients had reduced anti-RBD fucosylation compared with both anti-NA and anti-HA IgGs from influenza virus patients (Figure 3). To determine whether respiratory viral infections necessarily trigger a change in antibody glycosylation, we characterized anti-HA and anti-NA IgGs from the influenza virus-infected patients prior to, and at multiple timepoints after infection and observed that no significant changes in Fc glycosylation that were triggered by infection (Figure S3).

The abundance of fucosylation on anti-RBD lgG1 Fc from COVID-19 patients (red), seropositive children (light blue), total IgG from healthy adults (HC) (orange) and neuraminidase (NA) (green) or hemagglutinin (HA) (blue) IgG 1 from adult patients with symptomatic influenza virus infections. Data are representative of one mass-spectrometry-based experiment. P values were calculated using Wilcoxon rank sum tests. Violin plots show the distribution of sample values along with median (solid lines) and quartile (broken lines) values.
Despite evidence that antibodies can enhance infection by many viruses in vitro, these systems are rarely matched with evidence that antibodies promote viral infections or exacerbate the severity of viral diseases in humans. Dengue virus infections remain one of the few examples where there is compelling evidence that IgG antibodies can exacerbate the severity of disease during infection, including specific antibody modifications that are present in people with more severe dengue disease [9, 10, 22–26]. The present findings related to antibodies in SARS-CoV-2 infections show that a specific IgG Fc structure, reduced core fucosylation, correlates with COVID-19 disease. This Fc structure is enriched when compared to IgGs in SARS-CoV-2 seropositive children or in adults with symptomatic influenza virus infections. One important question to address in future studies will be whether core fucosylation is reduced prior to infection in individuals who are susceptible to COVID-19 disease or whether this modification is triggered by infection itself. If core fucosylation is reduced prior to infection, this could be a marker of susceptibility to COVID-19. That influenza infection does not trigger reduced core fucosylation of antibodies shows that respiratory viral infections do not necessarily trigger this antibody modification. Because reduced core-fucosylation of IgG1 antibodies enhances affinity of immune complexes for the activating FcγRIIIa, a second important question to address is whether the FcγRIIIa pathway may modulate the severity of SARS-CoV-2 infections. While the data presented here suggest that antibody effector function is important in COVID-19 disease, they do not indicate that the disease is necessarily a consequence of an enhancing process by antibodies. The FcγRIIIa pathway may contribute to killing and clearance of infected cells, promote inflammatory cell activity and/or modulate cytokine production during SARS-CoV-2 infections, any of which could contribute to viral clearance and benefit the host. Indeed, production of antibodies with reduced Fc fucosylation could be beneficial in SARS-CoV-2 infections. Should future studies show that IgG-FcγRIIIa interactions are involved in exacerbating symptoms of COVID-19, this would be a clear pathway to target for novel therapeutics. In summary, these findings show an antibody correlate of COVID-19 disease and can be used to guide studies into the pathogenesis of COVID-19 disease.
Methods
Cloning, expression and protein purification
The hexahistidine-tagged SARS-CoV-2 RBD construct was a kind gift from Florian Krammer. The full length recombinant SARS-CoV-2 spike protein (residues 1−1208 (GenBank:MN908947)) construct was designed with the following modifications: two well-characterized proline substitutions (K986P and V987P) [27]; a four amino acid substitution to remove the furin cleavage site (RRAR → GSAS) in order to stabilize the pre-fusion conformation [28]; a synthetic trimerization motif-the globular β-rich ‘foldon’ from T4 fibritin to promote oligomerization in lieu of the native trans-membrane (TM) domain; a human rhino virus 3C (HRV 3C) protease cleavage site; a C-terminal 8XHisTag. Mammalian codon-optimized gene fragments were synthesized (Integrated DNA Technologies, Inc.) and cloned using Gibson Assembly (New England BioLabs) into a CMV/R promoter driven mammalian expression vector between XbaI and BamHI restriction sites.
Both the constructs were transiently transfected into Expi293F cells (Thermo Fisher Scientific) as per the manufacturer’s recommendation. Briefly, Expi293F cells at a density of 3×106 viable cells/ml maintained in Expi293 expression medium (Thermo Fisher Scientific) were transfected with expression plasmids complexed with ExpiFectamine 293 transfection reagent. 18 hours posttransfection, the cells were supplemented with a cocktail of transfection enhancers. The cultures were incubated for four days, following which the culture supernatants were harvested by centrifugation for protein purification. The supernatants were incubated with phosphate-buffered saline (PBS (Gibco))-equilibrated Ni-nitriloacedic acid (NTA) resin (GE HealthCare) for 2h at 4°C under mild-mixing conditions to facilitate binding. The proteins were subsequently eluted using 500mM imidazole (in PBS, pH 7.4) under gravity flow. The eluted fractions were pooled, buffer exchanged into PBS (pH 7.4) and concentrated using Amicon Ultra centrifugal units (EMD Millipore) to a final concentration of ∼1mg/ml. Protein purity was assessed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). Size exclusion chromatography was used to determine the oligomeric state of the purified proteins under non-denaturing conditions at room temperature on a Superdex-200 analytical gel filtration column (GE HealthCare). For molecular weight estimations, the column was calibrated using broad range molecular weight markers (GE HealthCare).
ELISAs
Screening ELISA
A rapid, high-throughput screening ELISA was performed on a total of 1027 serum samples (225 PCR+ for SARS-CoV-2 and 802 pediatric samples) to test seropositivity following a modified version of a protocol described previously. Briefly, round bottom 96 well plates (Immunolon 2HB (Thermo Scientific)) plates were coated with 50μl of RBD at 2μg/ml in PBS for 1h at room temperature (RT). Next, the plates were blocked for an hour with 3% non-fat milk in PBS with 0.1% Tween 20 (PBST). All serum samples from COVID-19 patients, the pediatric cohort and the negative controls were heated at 56°C for 1h, aliquoted and stored at –80°C. For the first round of screening, all samples were diluted 1:50 in 1% non-fat milk in PBST. 50μl of the diluted sera was added to each well and incubated for 2h at RT. Following primary incubation with the sera, 50μl of 1:5000 diluted horse radish peroxidase (HRP) conjugated goat anti-Human Ig Fab (Southern Biotech) was added and incubated for 1h at RT. The plates were developed by adding 50ul/well of the chromogenic substrate 3,3′,5,5′-tetramethylbenzidine (TMB) solution (Millipore Sigma). The reaction was stopped with 0.2N sulphuric acid (Sigma) and absorbance was measured at 450nm (SPECTRAmax 250, Molecular Devices). The plates were washed 5 times with PBST between each step and an additional wash with PBS was done before developing the plates. Samples were considered seropositive against RBD if their absorbance value was greater than the mean plus four standard deviation (SD) of all negative controls (n=130).
Validation ELISA of seropositive pediatric samples
The serum samples from the pediatric cohort, which showed seropositivity against RBD were validated by a second round of screening against the full-length SARS-Cov-2 spike protein (S). As described above, plates were coated with 50ul of 2ug/ml S protein in PBS. Following blocking and wash, the plates were incubated with a 5-fold dilution series of RBD positive sera (50μl) starting at 1:50 for 2h at room temperature. All the subsequent steps were followed as described above. The sample was considered positive if the absorbance value at 1:250 serum dilution was higher than mean plus four SD of the negative controls.
Isotyping by ELISA
All the PCR+ samples which were positive by the rapid screen and pediatric samples which were positive after two rounds of screening were isotyped and titered against RBD. Sera were diluted 5-fold starting at 1:50 and ELISAs were performed as described above. The various secondary antibodies used for isotyping were 1:5000 dilutions of HRP-conjugated Goat Anti-Human IgG Fc (Southern Biotech), Mouse Anti-Human IgM (Southern Biotech) and Goat Anti-Human IgA Fc (Southern Biotech).
CD16a ELISA
Human recombinant CD16a (Sino Biological) was immobilized at 3ug/ml (50ul/well) in PBS at 4°C overnight, followed by an hour of blocking with 3% non-fat milk in PBST. 50ul of 1:50 diluted sera from pediatric seropositive (n=11), PCR+ and seropositive (n=22) and healthy controls (n=5) were added to each well and incubated for 2h at 37°C. Subsequently the plates were incubated for 1h at 37°C with 1:5000 dilution of HRP-conjugated Goat Anti-Human IgG Fc (Southern Biotech) secondary antibody, developed as described previously with TMB and absorbance was recorded at 450nm.
Clinical cohorts and samples
Remnant sera from pediatric subjects and from PCR+ COVID-19 patients were obtained from Kaiser Permanente Northern California. The sera were collected for a variety of clinical tests at one of 75 distinct hospitals or outpatient clinics across 17 counties in Northern California between March 30th, 2020 to April 19th, 2020. Additional serum samples from PCR+ COVID-19 patients were from the Stanford ICU Biobank (protocol #28205)[29]. Characterization of these samples was performed under a protocol approved by the Institutional Review Board of Stanford University (protocol #55718). Samples from the controlled influenza A virus challenge study have been previously described [21]. The study (ClinicalTrials.gov NCT01646138) was approved by the National Institute of Allergy and Infectious Diseases institutional review board and underwent an ethics review by the NIH Department of Bioethics prior to being conducted. The study was conducted in accordance with the provisions of the Declaration of Helsinki and Good Clinical Practice guidelines.
Samples from people with seasonal coronavirus infections were collected at the University of Chicago. Samples were de-identified serums of healthcare workers that had respiratory illnesses, were swabbed, and tested positive for common cold Corona virus infections in 2019 (U. Chicago protocol # 09–043-A).
Historical controls and healthy controls: 30 samples from a US cohort was enrolled at the Rockefeller University Hospital in New York City in 2012 in accordance with a protocol approved by the Institutional Review Board of Rockefeller University (protocol #TWA-0804), in compliance with guidelines of the International Conference on Harmonization Good Clinical Practice guidelines, and was registered on www.clinicaltrials.gov (NCT01967238). Blood samples were drawn from healthy adult volunteers between the ages of 18–64. 50 samples were obtained from a Ugandan cohort of women and children enrolled in PROMOTE (NCT 02163447), a randomized clinical trial of novel antimalarial chemoprevention regimens in Eastern Uganda (PMID 30016328). The study was approved by the Institutional Review Boards of the Makerere University School of Biomedical Sciences, the Uganda National Council for Science and Technology, and the University of California San Francisco. Written informed consent was obtained from all study participants. 50 samples were obtained from children under 18 years of age enrolled in a study of acute febrile illness in Nepal. The study was approved by the Nepal Health Research Council, Kathmandu University Institutional Review Board, and Stanford University Institutional Review Board (protocol #29992).
IgG Fc glycan and IgG subclass analysis
Methods for relative quantification of Fc Glycans and IgG subclasses have been previously described[9, 16]. Briefly, IgGs were isolated from serum by protein G purification. Antigen-specific IgGs were isolated on NHS agarose resin (ThermoFisher; 26196) coupled to the protein of interest. Following tryptic digestion of purified IgG bound to antigen-coated beads, nanoLCMS/MS analysis for characterization of glycosylation sites was performed on an UltiMate3000 nanoLC (Dionex) coupled with a hybrid triple quadrupole linear ion trap mass spectrometer, the 4000 Q Trap (SCIEX). MS data acquisition was performed using Analyst 1.6.1 software (SCIEX) for precursor ion scan triggered information dependent acquisition (IDA) analysis for initial discovery-based identification.
For quantitative analysis of the glycoforms at the N297 site of IgG1, multiple-reaction monitoring (MRM) analysis for selected target glycopeptide was applied using the nanoLC-4000 Q Trap platform to the samples which had been digested with trypsin. The m/z of 4-charged ions for all different glycoforms as Q1 and the fragment ion at m/z 366.1 as Q3 for each of transition pairs were used for MRM assays. A native IgGs tryptic peptide (131-GTLVTVSSASTK-142) with transition pair of, 575.9+2/780.4 was used as a reference peptide for normalization. IgG subclass distribution was quantitatively determined by nanoLC-MRM analysis of tryptic peptides following removal of glycans from purified IgGs with PNGase F. Here the m/z value of fragment ions for monitoring transition pairs was always larger than that of their precursor ions to enhance the selectivity for unmodified targeted peptides and the reference peptide. All raw MRM data was processed using MultiQuant 2.1.1 (SCIEX). All MRM peak areas were automatically integrated and inspected manually. In the case where the automatic peak integration by MultiQuant failed, manual integration was performed using the MultiQuant software.
Binding affinity measurements using biolayer interferometry (BLI)
Total IgGs were purified from sera pooled from pediatric seropositive, PCR+ seropositive and healthy controls using Protein G beads (n=7 samples per pool). The binding affinity of IgGs from the various groups was determined by biolayer interferometry (BLI) using an OctetQK instrument (Pall ForteBio). Human recombinant CD16a (Sino Biological) was captured onto the amine reactive second-generation (AR2G) biosensors using the amine reactive second-generation reagent kit (Pall ForteBio). The CD16a bound sensors were dipped into a concentration series (3.33μM, 1.7μM, 0.832μM and 0.33μM) of IgGs in PBST to determine the binding kinetics. An equal number of unliganded sensors biosensors dipped into the analytes served as controls for referencing.
The traces were processed using ForteBio Data Analysis Software version 8.0.3.5 and corrected for non-specific binding. The data was fitted globally to a simple 1:1 Langmuir interaction model.
Statistical analysis
All data were analyzed with GraphPad Prism 8.0 software. Investigators were blinded to study subjects diagnoses during screening; COVID-19 patients and children were not known by investigators at the time of ELISA screening for RBD reactivity of serum or by investigators involved in relative quantitation of Fc glycoforms and IgG subclasses by mass spectrometry.
Data Availability
All raw data are available from the corresponding author on reasonable request.
Author contributions
T.T.W, J.P., S.C., K.E., M.J.M, J.K.T. conceived of and/or designed experiments. T.T.W, J.P., S.C., K.E., R.S., S.Z., A.B., M.X., J.G., C.B., N.K., S.P., J.R.A., J.M.S, T.M., C.A.B., F.K., P.C.W, T.D.P., S.B. were involved in data acquisition and/or analysis and/or interpretation. S.C. and T.T.W wrote the manuscript.
Competing interests
The authors declare no competing interests.
Data availability
All raw data are available from the corresponding author on reasonable request.
Acknowledgments
Support was received from Stanford University, the Chan Zuckerberg Biohub and the Searle Scholars Program. Research reported in this publication was supported by Fast Grants, CEND COVID Catalyst Fund, the NIH/NIAID (U19AI111825, R01AI139119 and NIH Contract 75N93019C00051, Sinai-Emory Multi-Institutional CIVIC) and by the Rockefeller University Center for Clinical and Translational Science Grant # UL1 TR001866. The influenza A virus challenge study was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases (NIAID). The authors thank Celeste Kopel for excellent technical support.




{kind=link}
{kind=link}
{kind=link}