
Abstract
Rapid, reliable, and widespread testing is required to curtail the ongoing COVID-19 pandemic. Current gold standard diagnostic assays are hampered by supply shortages in critical reagents including nasal swabs, RNA extraction kits, personal protective equipment (PPE), instrumentation, and labor. Here we present an approach to overcome these challenges with the development of a rapid colorimetric assay using reverse-transcription loop-mediated isothermal amplification (RT-LAMP) optimized on human saliva samples without an RNA purification step. We describe our optimizations of the LAMP reaction and saliva pre-treatment protocols that enabled rapid and sensitive detection of < 102 viral genomes per reaction in contrived saliva controls. We also observed high performance of this assay on a limited number of clinical saliva samples. While thorough validation on additional clinical samples will be needed before such an assay can be widely used, these preliminary results demonstrate a promising approach to overcome the current bottlenecks limiting widespread testing.
Introduction
Establishing rapid and widespread testing for coronavirus disease 2019 (COVID-19) is essential to containing the pandemic and reopening society. The current gold standard test measures viral nucleic acids extracted from clinical swabs by quantitative reverse transcription polymerase chain reaction (qRT-PCR). This assay requires trained medical personnel, specialized instrumentation, supply-limited reagents, and significant technical labor. Isothermal nucleic acid amplification tests are an alternative to conventional PCR methods that do not require expensive instruments or trained personnel to perform the reaction or quantify the results. Specifically, loop-mediated isothermal amplification (LAMP) with simultaneous reverse-transcription (RT-LAMP) allows for rapid and sensitive detection of nucleic acids within one hour in an easily interpretable colorimetric assay that requires only a heat source1,2.
Several groups around the world are currently developing LAMP-based protocols for the detection of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the virus causing COVID-193–7. The sensitivity of LAMP compares well to the limit-of-detection of qRT-PCR on purified RNA samples, and may achieve higher sensitivity on crude clinical samples5. The robustness of the LAMP Bst polymerase to PCR inhibitors makes it especially well-suited and widely used for pathogen detection in unpurified samples8. This confers a major potential advantage over current testing protocols as it enables skipping the cost-, labor-, and reagent-consuming step of RNA extraction.
Saliva is a promising sample for expanding and facilitating testing due to the ease, safety, and non-invasive nature of its collection and its relatively high viral load9,10. Recognizing these benefits, the FDA approved a saliva collection and preservation device for downstream COVID-19 testing. Direct comparison of saliva to nasopharyngeal (NP) swabs from the same individuals revealed that saliva samples provided more consistent and sensitive results for COVID-19 detection11. These saliva-based methods, however, still employ RNA extraction followed by qRT-PCR.
Here, we sought to establish and optimize a simple LAMP-based assay for the qualitative detection of SARS-CoV-2 virus directly from saliva without an RNA extraction step.
Results
LAMP Primer Screening
To develop our assay, we first compared the performance of five sets of recently developed LAMP primer sets targeting different regions of the SARS-CoV-2 genome3–6. We used a commercially available NEB colorimetric enzyme mix to perform LAMP reactions on quantitative in vitro transcribed RNA standards corresponding to regions targeted by LAMP primers12. Of these, the NEB Gene N-A3 and Lamb et al.4 primers targeting the nucleocapsid (Gene N) and Orf1ab regions respectively had the highest sensitivity and lowest rates of false positives in the water-only control (Supplementary Figure 1). Demonstrating specificity to SARS-CoV-2, these primers had no cross-reactivity with MERS coronavirus controls. These primer sets were prioritized for further testing.
Reaction Optimization in Simulated Samples
Next, we validated these primer sets on both RNA standards and heat-inactivated viral particles spiked into water or human saliva to simulate clinical samples (Figure 1A). Across both sets of LAMP primers, and for both RNA and particles, saliva strongly inhibited LAMP detection of SARS-CoV-2 compared to water (Fig. 1B). Particles were weakly and inconsistently detected in saliva whereas their detection in water was on par with detection of RNA (Fig. 1C). This suggests the presence of an inhibitor in saliva rather than an inaccessibility of particle-associated RNA. We observed time sensitivity of the colorimetric assay especially from saliva, with many samples tinting yellow after prolonged incubation in the LAMP reaction (longer than 40 minutes). We found that a 30-minute incubation provided a more reliable readout.
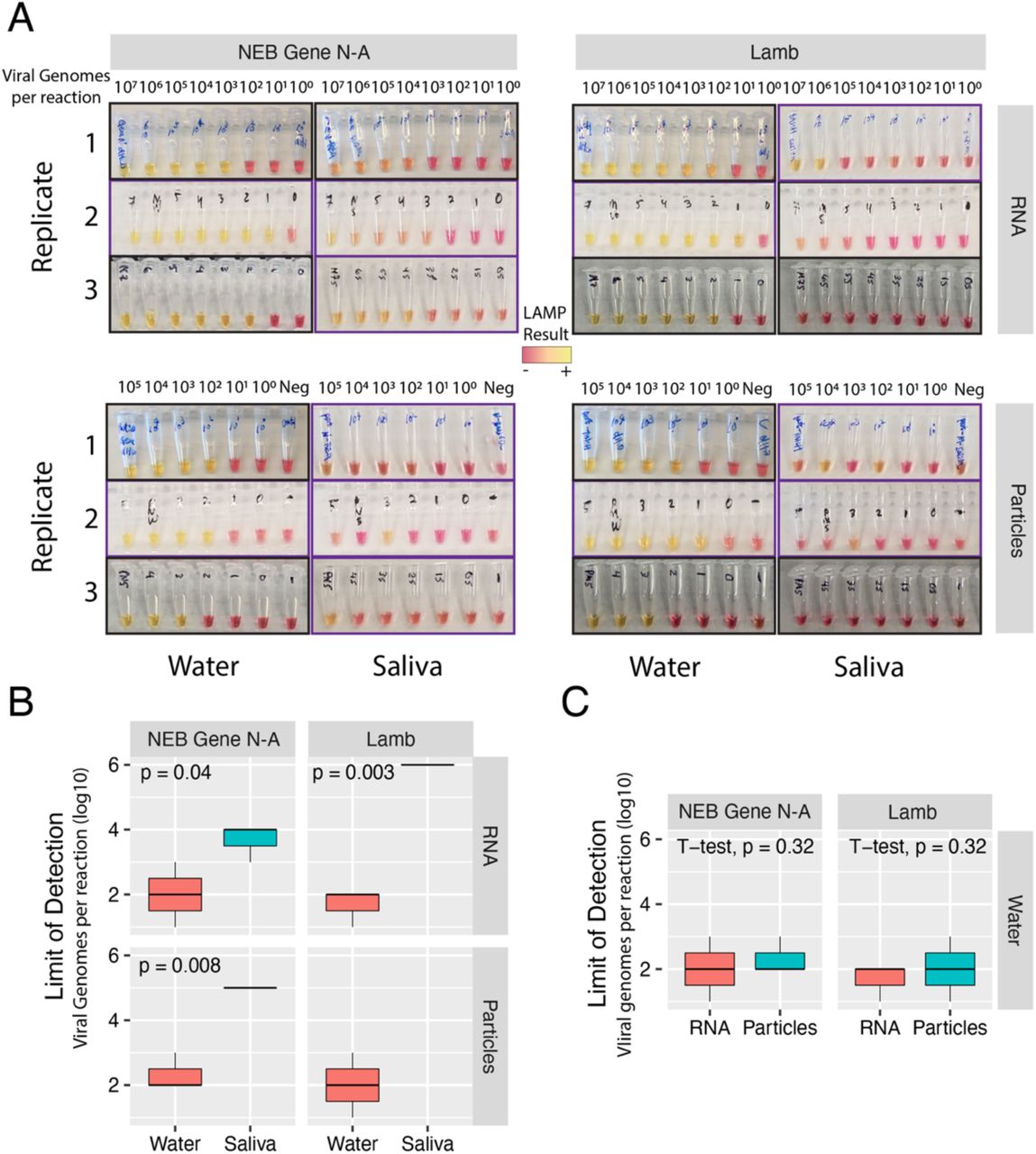
A) LAMP reactions of RNA or particles in water or saliva, amplified with NEB Gene N-A primers or Lamb et al primers as indicated. Independent biological triplicates are shown for each experiment. Values indicate number of viral genome equivalents per reaction. Purple-framed boxes indicate 30-minute reactions. Black-framed boxes indicate 1-hour reactions. B) Quantification and comparison of approximate Limits of Detection (viral genome equivalents per reaction) were compared by RNA type, water or saliva, and primer sets. Limit of Detection was recorded as the lowest value with a clear colorimetric change from magenta to yellow. Inconclusive or undetected values were recorded as Not Detected. Quantification and comparison of approximate Limits of Detection (viral genome equivalents per reaction) were compared by RNA type, water or saliva, and primer sets. P-values indicate one-sided t-tests (saliva greater than water). Inconclusive or non-detected values were excluded.
To neutralize or otherwise reduce inhibitors in human saliva, we tested several approaches that have been demonstrated to improve viral RNA detection in crude samples including saliva13-17. We tested simple dilution of particle-containing saliva into water, and various heat and chemical treatments. First, we found that dilution of saliva into water enabled sensitive detection of SARS-CoV-2 particles using LAMP (Figure 2A, top). A heat treatment of 55°C for 15 minutes followed by 98°C for 5 minutes further improved LAMP sensitivity (Fig. 2A, middle). Identical heating steps plus the addition of proteinase K increased LAMP sensitivity relative to dilution alone (Fig. 2A, bottom) but not markedly more than heat treatment. Importantly, we found that the combined heating steps above or proteinase K treatment improved SARS-CoV-2 particle detection in undiluted human saliva samples (Fig. 2B) and conferred a consistent Limit of Detection on the order of 102 particles per reaction. We experimented with additional heat and chemical pretreatments including the HUDSON protocol (heating unextracted diagnostic samples to obliterate nucleases)14 and various detergents, but all of these conditions decreased assay sensitivity or interfered with colorimetry (Supplementary Figure 2A-C). We also varied the amount of crude sample input to the LAMP reaction. We found that adding up to 8 μL of direct saliva was compatible with the assay but increased volume did not improve sensitivity (Supp. Fig. 2D).

A) Dilution of particle-containing saliva into water improved LAMP detection by at least two orders of magnitude from undetectable to ~103 particles per reaction. Heat treatment and heat treatment plus proteinase K further increased LAMP sensitivity to ~102 viral genome equivalents per reaction (p < 0.1, one-sided t-tests compared to dilution alone). *Replicate 3 used Lamb et al. primers but gave nearly identical results to NEB Gene N-A primers. B) Heat treatment or heat treatment plus proteinase K treatments increased LAMP sensitivity from undetectable to ~102 viral genome equivalents in undiluted saliva. All reactions are purple-framed to indicate 30-minute reactions. W = water, S = saliva.
Multiplexing LAMP Primer Sets
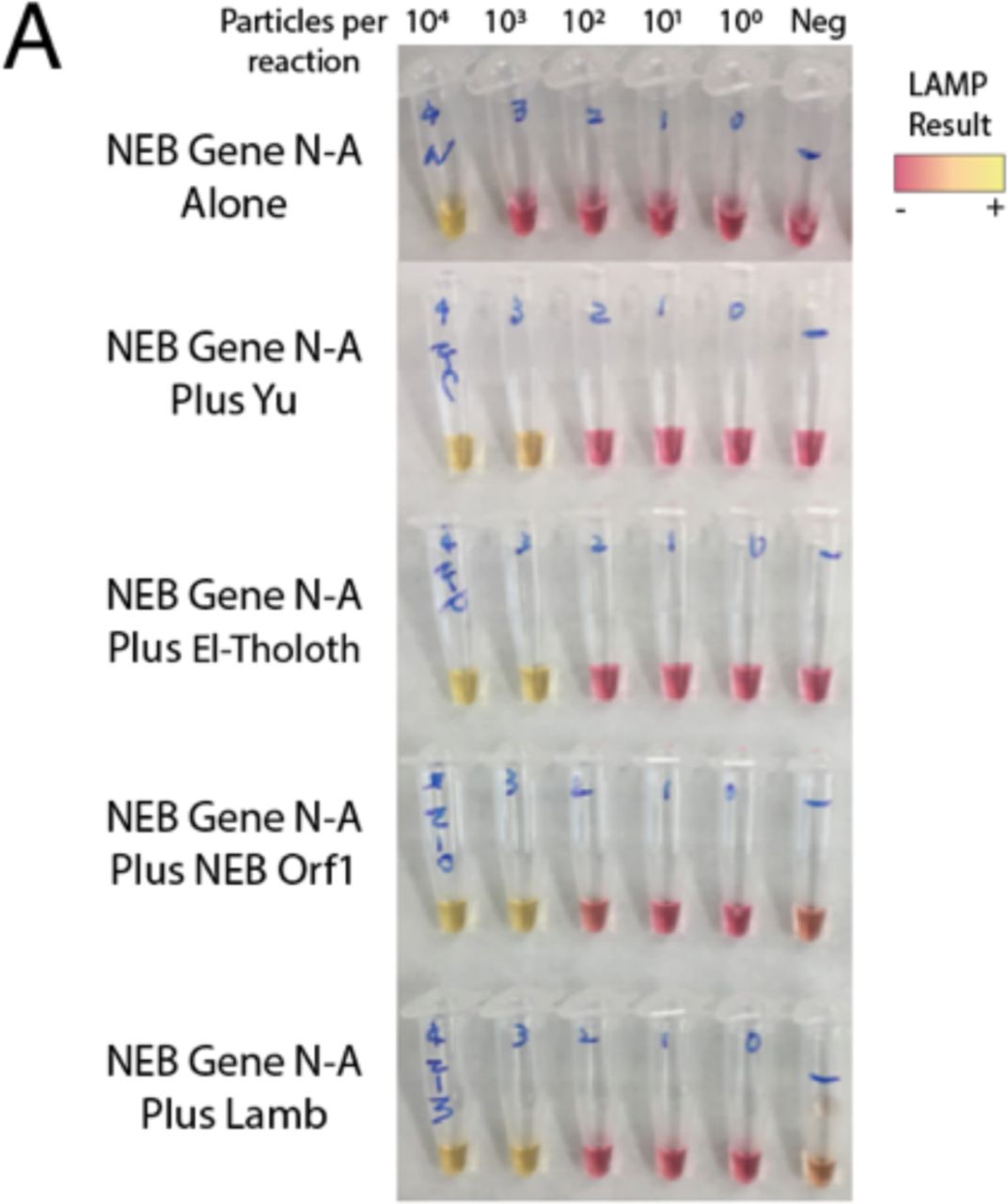
To further improve the accuracy of our assay, we sought to multiplex LAMP primer sets in a single reaction. Combining primers can potentially increase sensitivity through additive signals of simultaneous amplification reactions18,19. Including multiple primer sets will also confer diagnostic robustness against mutations that arise in the SARS-CoV-2 genome20. Non-specific primer interactions, however, could result in potential false positives. We compared pairwise combinations of NEB Gene N-A primers with the other four primer sets targeting various regions across the SARS-CoV-2 genome. Encouragingly, all pairs of primer sets outperformed the NEB Gene N-A primer set alone, with no apparent increase in spurious background amplification (Supplementary Figure 3).
We next tested whether multiplexing primer sets could improve signal detection in untreated and heat and chemical treated particle-containing saliva (Figure 3A). As before, we found that heat treatment (55° for 15 minutes, 98° for 3 minutes) alone gave a marked improvement in SARS-CoV-2 particle detection from saliva (Fig. 3B, p < 1e-5, two-sided t-test). This effect was consistent across all primer sets. The same heat treatment plus proteinase K further improved assay sensitivity compared to heat alone (p < 0.003, two-sided t-test). Multiplexed primer sets slightly improved the sensitivity of the assay, pushing the limit of detection to the order of ~101 particles per reaction. At this sensitivity, the multiplexed LAMP assay would detect the vast majority of COVID positive samples based on reported saliva viral loads (median ~102-103 per uL)10,11. As viral loads peak around day zero of symptom onset, LAMP would have the most accuracy at this critical timepoint21.
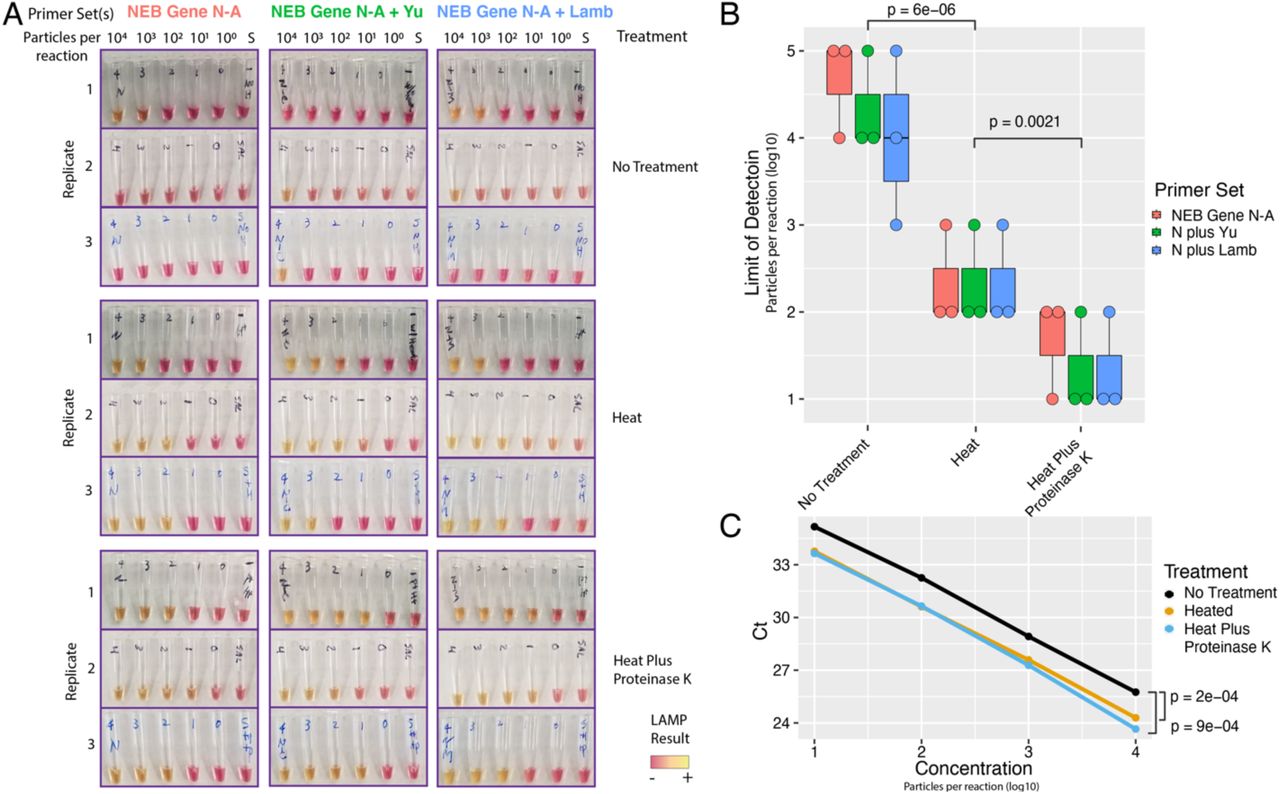
A) LAMP reactions using NEB Gene N-A primers alone or in combination with Yu et al. or Lamb et al. primers are shown. S = negative control saliva. Viral particles per reaction are indicated. B) Saliva pre-treatments greatly improve LAMP sensitivity. Heat treatment improves LOD (p = 6e-6, t-test, two-tailed vs ‘Untreated’). Proteinase K treatment further improves heat treatment (p = 0.002, t-test, two-tailed vs ‘Heat’). Multiplexed primers may slightly improve Limit of Detection to ~ 101 particles / reaction. C) qRT-PCR on crude lysate (NEB Gene N-A reactions) showed similar increase in sensitivity with either heat or proteinase K treatment (p < 1e-3 for either treatment, 2-tailed paired t-test). Proteinase K did not further improve qRT-PCR sensitivity. N = NEB Gene N-A.
To benchmark the performance of our extraction-free protocol on qRT-PCR, we performed qRT-PCR using the CDC Gene N1 probe set directly on untreated and treated simulated saliva samples. We found that qRT-PCR had similar sensitivity to LAMP on crude samples, reliably detecting SARS-CoV-2 in all samples down to 101 particles per reaction (Fig. 3C). 10° particles were not reliably detected in this assay. We observed strong improvements in cycle thresholds (Ct) using either heat alone or heat plus proteinase K (p < 1e-3, two-tailed paired t-tests). These results provide further evidence that saliva pretreatment can significantly increase the sensitivity of viral RNA detection.
Validation on Clinical Samples
We obtained saliva samples collected at day zero of hospital admission from six presumptive COVID-19 positive individuals. One sample was excluded because its viscosity prevented pipetting. The other five were aliquoted into 3 tubes for either no treatment, heat inactivation (55° for 15 minutes, 98° for 3 minutes), or the same heat treatment plus proteinase K. Following pretreatment, the RT-LAMP reaction was performed with the NEB Gene N-A primers. Photographs were taken at 10, 20 and 30 minutes to track colorimetric shifts over time (Figure 4A). An aliquot of 104 viral particles in saliva was used as a positive control. After 30 minutes, 4 of the five samples were clearly positive in the heat plus proteinase K treated samples (Fig. 4A, bottom right). Positive and negative controls were positive and negative in all reactions at the 30-minute timepoint, allowing an interpretable readout of the assay. Untreated samples showed positivity for samples 3-5, and heat-alone indicated clear positivity for samples 2-4. These differences may reflect altered ratios of free viral RNA to particle-associated RNA in these samples, which could be differentially affected by our heat treatment. Proteinase K treatment both protects free RNA by inactivating nucleases, and releases particle-bound RNA. Incorporating quantitative colorimetric decomposition and analysis will enhance sensitivity and interpretability of borderline samples in future experiments22.

A) Five presumptive COVID positive saliva samples were subjected to the indicated pretreatment protocol followed by RT-LAMP. Heat = 55° for 15 minutes, 98° for 3 minutes, with or without proteinase K. Photographs for timepoints 10- and 30-minutes are shown. By 30 minutes, all positive controls are positive (yellow), and negative controls remained negative (magenta). Heat plus Proteinase K treatment indicated 4/5 positives. B) qRT-PCR performed directly on crude pre-treated saliva samples was qualitatively concordant with the LAMP assay. Samples 2-5 are positive (Ct < 40). Sample 1 and negative control gave no signal. C) Pretreatment protocols enhance sensitivity of qPCR (two-sided paired t-test, ** = p < 0.01, *** p < 0.001). ProK, proteinase K. Ct, cycle threshold.
We did not have corresponding gold-standard qRT-PCR results from these samples. Instead, we performed qRT-PCR with the CDC N1 probe directly on the untreated and treated samples, along with our positive and negative controls. qRT-PCR results were qualitatively concordant with the RT-LAMP results, identifying 4 clear positives (Samples 2-5, Ct < 40, Fig. 4B). Sample #1 tested negative in all LAMP treatments and qRT-PCR, and an attempt to purify RNA from this saliva sample was unsuccessful. This may indicate RNA degradation prior to LAMP and qRT-PCR. Further work is needed to establish best practices for upstream handling of saliva specimens. Toward this end, we have shown that the LAMP reaction is compatible with samples in TE buffer (10 mM Tris, 0.1 mM EDTA), diluted HUDSON buffer (2.5 mM TCEP, 0.5 mM EDTA), and RNAsecure (Supplementary Figure 4A).
As with LAMP, our pre-treatment regimens significantly improved nucleic acid detection by qRT-PCR (Fig. 4C). Heat treatment (55° for 15 minutes, 98° for 3 minutes) with and without proteinase K outperformed untreated samples (p < 0.01 and p < 0.001 respectively). Proteinase K did not further significantly improve sensitivity compared to heat alone. Extrapolation of viral loads in clinical saliva samples by comparing to our quantitative positive control yielded estimates of 1.4 × 101 − 9.8 × 102 particles per μL and demonstrates the sensitivity of our assay on real clinical samples. Further validation and optimization on additional positive and negative saliva specimens are necessary for clinical assay deployment.
Establishing a High-throughput Quantitative Assay
To enable significant scale-up of testing capacity using the LAMP assay on human saliva, we adapted our protocol to a 96-well plate format. Spectrophotometric plate scanning before and after the assay provided an unbiased, quantitative interpretation. Initial plate scanning was implemented to normalize for baseline differences induced by variation in pH across human saliva samples. Heat treatment (55° for 15 minutes, 98° for 3 minutes) with and without proteinase K enabled sensitive detection of viral particles in human saliva samples down to 102 particles per reaction, with some detection at 101 and even 100 particles per reaction (Figure 5A). This assay was quantitative over at least four orders of magnitude. Quantification of the limit-of-detection and assay validation on clinical specimens are ongoing.
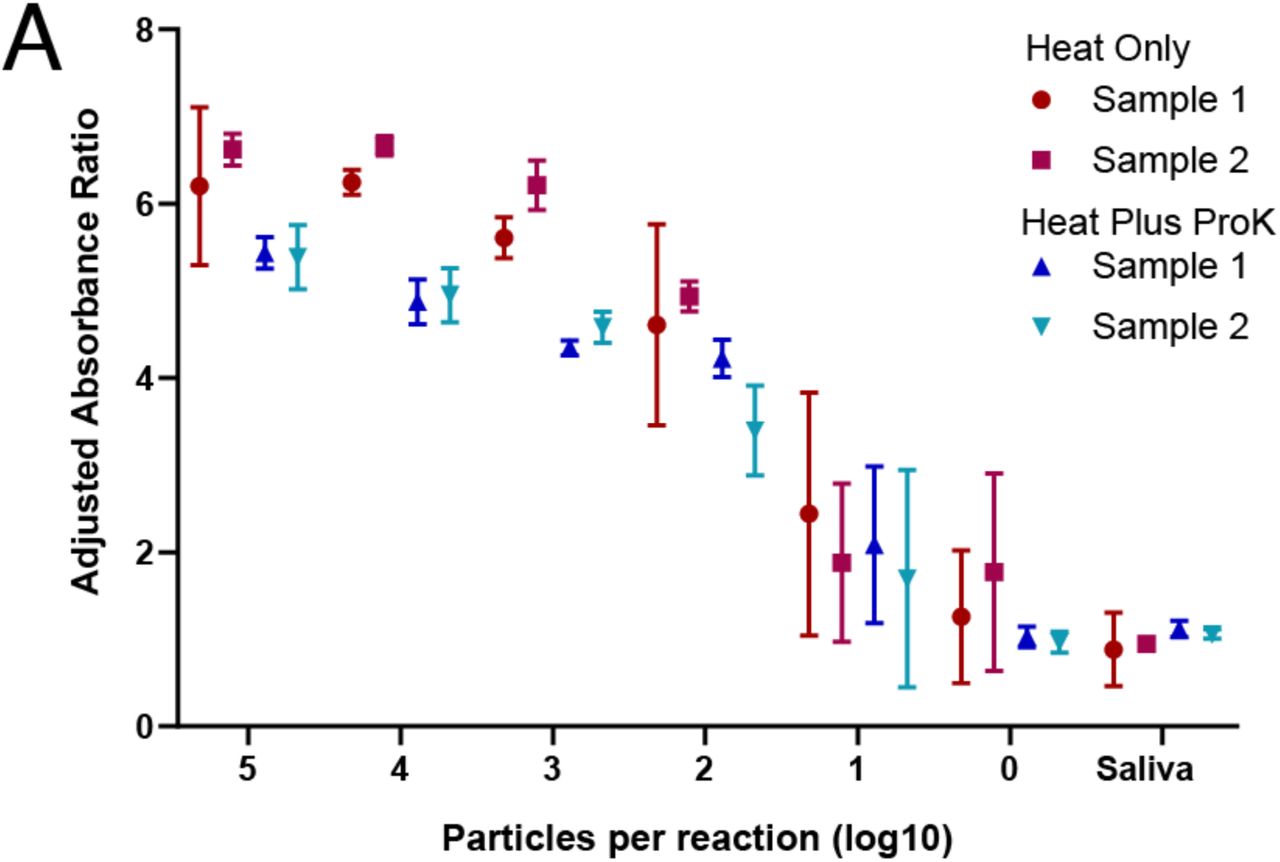
A) RT-LAMP assay was adapted to a high-throughput 96-well plate format with a quantitative absorbance readout, achieving a limit of detection < 102 particles per reaction from saliva samples. Absorbance for 430 nM (yellow) and 560 nM (red) wavelengths was measured before and after the LAMP reaction and normalized to negative controls. Heat = 55° for 15 minutes, 98° for 3 minutes, with or without proteinase K (proK). Two biological replicates were each run in triplicate.
Improving Compatibility with Point-of-Care Testing
Isothermal LAMP is well-suited to point-of-care testing because it requires only a heat-source. We sought to avoid the 98 °C heating step to make the saliva pre-treatment compatible with a single isothermal heat source. Whereas a mild heat treatment consisting of 50 °C for 5 minutes and 64° for 5 minutes did not enable sensitive detection of SARS-CoV-2 in undiluted saliva (Sup. Figure 2B), a 64° C treatment for 15 minutes of saliva that had been diluted 1:1 in either TE (10 mM Tris, 0.1 mM EDTA) or dilute HUDSON buffer (2.5 mM TCEP, 0.5 mM EDTA) improved detection (Sup. Fig. 4B). Addition of RNAsecure, a non-enzymatic ribonuclease inhibitor that does not require heat-inactivation, boosted assay sensitivity to 102 viral particles per reaction (Sup. Fig 4B). SARS-CoV-2 in samples treated with a thermolabile version of proteinase K (inactivated by incubation at 65° for 10 minutes) was not detected in a sensitive fashion (Sup. Fig 4C). Finally, we found that guanidine hydrochloride (40 mM) was compatible with the LAMP reaction and that both RNAsecure and primer multiplexing provided further enhancements to sensitivity (Sup. 4D). These pretreatment methods will enable the use of saliva in LAMP reactions using a single heat step, simplifying point-of-care testing.
Discussion
Our proposed approach combines three promising avenues to enable rapid and widespread COVID-19 detection: 1) colorimetric RT-LAMP, 2) self-collected saliva specimens, and 3) compatibility with crude saliva samples without RNA-extraction. This approach solves two major bottlenecks in massively scaling up COVID-19 nucleic acid testing: sample acquisition and RNA extraction, and it enables test results in less than an hour. Viral shedding likely begins several days prior to onset of any symptoms and viral load peaks during this period21. Colorimetric RT-LAMP directly on saliva would enable rapid and frequent testing of pre- or asymptomatic carriers, enabling their isolation prior to unwitting viral transmission. Such testing is critical to curtailing the ongoing pandemic.
Due to its ease of use, rapid amplification of nucleic acids, high specificity arising from the use of six primers, and high tolerance of reaction inhibitors8, RT-LAMP has been widely used for pathogen detection. Sensitive diagnostic assays have been developed for viruses including ZIka19,23 and such assays are being developed for SARS-CoV-2 by several groups including ours3–7,18,24–28. Speed, cost, turnaround time, and a simple colorimetric readout make RT-LAMP an effective solution to ramping up testing. Further, because it does not require specialized equipment or training for performing or interpreting the assay, RT-LAMP is especially well-suited for point-of-care detection.
Sample acquisition is currently limited by the reliance on NP swabs, which need to be carefully performed by a trained health-care worker and require the use of PPE. Mid-nasal swabs are a promising alternative to NP swabs because they can be self-administered, and contain high viral loads29–31. However, due to potential swab shortages, we instead focused on expectorated saliva due to its ease of collection and high viral load11. Our work adds to the growing body of evidence that saliva will facilitate the adoption of widespread testing.
Several groups are optimizing workarounds to avoid the RNA extraction step for PCR based SARS-CoV-2 testing while maintaining sensitivity13,15,17,29. Here, we have demonstrated a variety of saliva pre-treatment protocols that enable sensitive detection of SARS-CoV-2 by both RT-LAMP and qRT-PCR. RNA in saliva likely comprises a mixture of free and particle-associated RNA. Capsid release of RNA and nuclease inactivation by heat or chemicals may damage free viral RNA, so a careful balance must be achieved to maximize sensitivity. Despite requiring brief inactivation at 98°C, proteinase K treatment to release viral RNA and inactivate ribonucleases and other inhibitors in saliva worked well to increase sensitivity in both RT-LAMP and qRT-PCR. Alternatively, TE buffer, guanidine, HUDSON buffer, and RNAsecure allowed us to perform a heat step isothermal with the LAMP reaction conditions while maintaining high assay sensitivity. This version of the protocol is compatible with point-of-care testing. Our proof-of-concept validation on actual clinical samples suggests high sensitivity of the current assay to detect SARS-CoV-2 directly from treated saliva. Further optimizations may improve assay sensitivity and robustness.
Daily screening of employees, especially health-care workers, is feasible with this approach due to its rapid turnaround, low complexity, and low cost. As viral loads are correlated with transmission, only weakly infectious carriers risk false negative results which is also true for the current gold standard. Colorimetric RT-LAMP on saliva has broad potential to increase COVID-19 screening speed and capacity, as demonstrated by our adaptation of this assay to a plate-based format. Similar approaches by others have now been validated on clinical samples, deployed for disease surveillance, and adapted for home testing and point-of-care use27,28.
In summary, we have optimized RT-LAMP reaction conditions to enable sensitive SARS-CoV-2 detection from unpurified saliva samples. This optimization overcomes the burden of RNA extraction reagent, need for sophisticated instrumentation, and the time and labor bottlenecks of the current gold standard nucleic acid-based tests. Our assay can be deployed as a point-of-care test or in a centralized laboratory facility. While non-exhaustive, our current optimizations have enabled reliable detection below ~102 viral genomes per reaction from simulated saliva samples. Using these conditions, we observed high performance of this assay on a limited number of clinical saliva specimens without RNA extraction.
Methods
LAMP Reactions
All LAMP reactions were performed following New England Biolab’s recommended protocol using WarmStart Colorimetric LAMP 2X Master Mix (NEB, Massachusetts USA, M1800L). 20 μL reactions with 10 μL LAMP master mix, 2 μL of 10X primer mix (2 μM F3 and B3, 16 μM Forward Inner Primer (FIP) and Backward Inner Primer (BIP), and 4 μM of Loop Forward (LF) and Loop Backward (LB) primers (25 or 100 nmol scale IDT), 5 μL nuclease-free water, and 3 μL samples. LAMP reactions were incubated at 65°C using BioRad DNA Engine thermocyclers for 30-60 minutes. Photographs were taken with cell phone cameras on samples laid on white sheets of paper.
SARS-CoV-2 Standards and Controls
In vitro transcribed RNA standards were prepared as described12. https://www.protocols.io/view/generation-of-sars-cov-2-rna-transcript-standards-bdv6i69e In brief, gBlocks (IDT) corresponding to SARS-CoV-2 regions targeted by LAMP primer sets were PCR amplified and in vitro transcribed using MEGAshortscript T7 Transcription Kit (ThermoFisher). RNA products were column-purified using the Monarch RNA Cleanup kit (NEB) and quantified using Qubit.
Heat inactivated SARS-CoV-2 particles were acquired from CDC through BEI Resources. Particle stock concentrations were 1.16 × 106 particles per μL. 10 μL particles were resuspended in 90 μL water or saliva to make 105 particles/ μL stock. Alternatively, 10 μL particles were added into 990 μL saliva to make 104 particles/ μL stocks which were individually aliquoted to reduce freeze-thaw.
DNA plasmid coronavirus controls corresponding to SARS-COV-2 and MERS were obtained from IDT as plasmid DNA solutions. nCoV-N control: Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome (GenBank: NC_045512.2). MERS control: Middle East respiratory syndrome-related coronavirus isolate KNIH/002_05_2015, complete genome (GenBank: MK796425.1)
Saliva Pretreatments
HUDSON (heating unextracted diagnostic samples to obliterate nucleases)14 was performed as described using TCEP (100 mM) and EDTA (1 mM) final concentrations. A mild heat treatment consisting of 50 °C for 5 minutes and 64° for 5 minutes was employed. Neutral pH TCEP was also investigated, but still caused an instant colorimetric shift in LAMP reactions. While diluted TCEP/EDTA was compatible with LAMP, its ability to inactivate nucleases or lyse viral particles remains to be validated.
Proteinase K from NEB (#P8107S) was added to saliva at 1/10 volume (5 μL in 50 μL saliva). 1:1 and 1:10 dilutions of Proteinase K into water or TE were also used. All concentrations were effective so 1:10 dilutions were used in ongoing experiments. We performed heat inactivation of Proteinase K for 5 minutes at 98°C. We also tested thermolabile proteinase K from NEB (P8111S). Samples were incubated at 37 °C for 10 minutes followed by proteinase K inactivation at 65 °C for 10 minutes.
RNAsecure (25X, ThermoFisher, AM7006) was added to some reactions at 1X.
During our optimizations, we observed that low levels of TE buffer were effective in preventing negative control samples from tinting yellow over prolonged LAMP reactions. A pulse spin in a microfuge improved the reliability of viral detection across various batches of saliva.
qRT-PCR
qRT-PCR reactions were performed according to CDC EUA guidelines using TaqPath 1-Step RT-qPCR Master Mix GC (Thermo A15300) and the nCoV-N1 probe form the 2019-nCoV RUO Kit (IDT). Reactions were performed on Quantstudio 3 and 6 Real-Time PCR systems (ThermoFisher). When using crude lysate as input, 3 μL was used as input to match the LAMP protocol.
Assay Scale-up
Assay scale-up was performed in a 96-well plate format (BioRad 96 well skirted PCR plate) with only minor modifications to the LAMP reaction. 4 μL of saliva samples were used in 25 μL total volume reactions. Heat treatment (55° for 15 minutes, 98° for 3 minutes) and proteinase K treatment were identical to single tube format. RNasin (Promega) was included in these samples. Samples were run in technical triplicate at each dilution.
Utilizing a BioTek Epoch microplate spectrophotometer, sample plates are scanned prior to heating, designated as the pre-read, to establish “background” due to variations in color related to individual saliva samples. After heating at 65 °C for 30 minutes, the plate was read a second time, designated as the post-read. Each read takes approximately 1 minute. The reader software returns endpoint absorbance for 430 nM and 560 nM wavelengths that measure positive and negative reaction results, respectively. Analytically, the ratio of 430 nM to 560 nM is computed for the pre- and post-read scans. For each well, the pre-read ratio is subtracted from the post-read ratio to establish a background subtracted value. The mean and standard deviation of the background subtracted negative controls (saliva + PBS) were computed from 12 wells across the plate. For each replicate, an adjusted absorbance ratio is computed by taking the ratio of each sample to the average negative control value (background subtracted sample ratio / background subtracted negative control ratio).
LAMP Primers
NEB_orf1a-A-F3 CTGCACCTCATGGTCATGTT
NEB_orf1a-A-B3 AGCTCGTCGCCTAAGTCAA
NEB_orf1a-A-FIP GAGGGACAAGGACACCAAGTGTATGGTTGAGCTGGTAGCAGA
NEB_orf1a-A-BIP CCAGTGGCTTACCGCAAGGTTTTAGATCGGCGCCGTAAC
NEB_orf1a-A-LF CCGTACTGAATGCCTTCGAGT
NEB_orf1a-A-LB TTCGTAAGAACGGTAATAAAGGAGC
NEB_geneN-A-F3 TGGCTACTACCGAAGAGCT
NEB_geneN-A-B3 TGCAGCATTGTTAGCAGGAT
NEB_geneN-A-FIP TCTGGCCCAGTTCCTAGGTAGTCCAGACGAATTCGTGGTGG
NEB_geneN-A-BIP AGACGGCATCATATGGGTTGCACGGGTGCCAATGTGATCT
NEB_geneN-A-LF GGACTGAGATCTTTCATTTTACCGT
NEB_geneN-A-LB ACTGAGGGAGCCTTGAATACA
El-Tholoth_orf1ab-F3 TGCTTCAGTCAGCTGATG
El-Tholoth_orf1ab-B3 TTAAATTGTCATCTTCGTCCTT
El-Tholoth_orf1ab-FIP TCAGTACTAGTGCCTGTGCCCACAATCGTTTTTAAACGGGT
El-Tholoth_orf1ab-BIP TCGTATACAGGGCTTTTGACATCTATCTTGGAAGCGACAACAA
El-Tholoth_orf1ab-LF CTGCACTTACACCGCAA
El-Tholoth_orf1ab-LB GTAGCTGGTTTTGCTAAATTCC
Lamb_F3 TCCAGATGAGGATGAAGAAGA
Lamb_B3 AGTCTGAACAACTGGTGTAAG
Lamb_FIP AGAGCAGCAGAAGTGGCACAGGTGATTGTGAAGAAGAAGAG
Lamb_BIP TCAACCTGAAGAAGAGCAAGAACTGATTGTCCTCACTGCC
Lamb_LF CTCATATTGAGTTGATGGCTCA
Lamb_LB ACAAACTGTTGGTCAACAAGAC
Yu_orf1ab-F3 CCACTAGAGGAGCTACTGTA
Yu_orf1ab-B3 TGACAAGCTACAACACGT
Yu_orf1ab-FIP AGGTGAGGGTTTTCTACATCACTATATTGGAACAAGCAAATTCTATGG
Yu_orf1ab-BIP ATGGGTTGGGATTATCCTAAATGTGTGCGAGCAAGAACAAGTG
Yu_orf1ab-LF CAGTTTTTAACATGTTGTGCCAACC
Yu_orf1ab-LB TAGAGCCATGCCTAACATGCT
Clinical Samples
Saliva samples were collected at day zero of hospital admission from six presumptive COVID-19 positive individuals. Saliva samples were diluted 1:1 in phosphate buffered saline to facilitate pipetting and then frozen. Samples were then thawed and heat-treated at 56°C for 30 minutes to inactivate the majority of live virus32. Samples were then re-frozen. Samples were thawed on ice prior to LAMP and qRT-PCR reactions.
Ethics Approval
Saliva collection was approved by the institutional review board at Washington University School of Medicine (WU350, IRB#202003085). Informed consent was obtained for all participant samples.
Data Availability
All relevant data are contained within the paper.
Author Contributions
MAL, XC, SJL, CSS, LCB, WJB developed and optimized the LAMP assay with significant intellectual contribution from MH, RDM, RSF, RDH, JM. Clinical samples were obtained and prepared by MAL, CCF, and RSF. Assay scale-up and quantitative read-out were implemented and analyzed by CSS, LCB, MH, and RDH. MAL analyzed the data and prepared the manuscript, with editing and revision by all authors.
Supplementary Figures
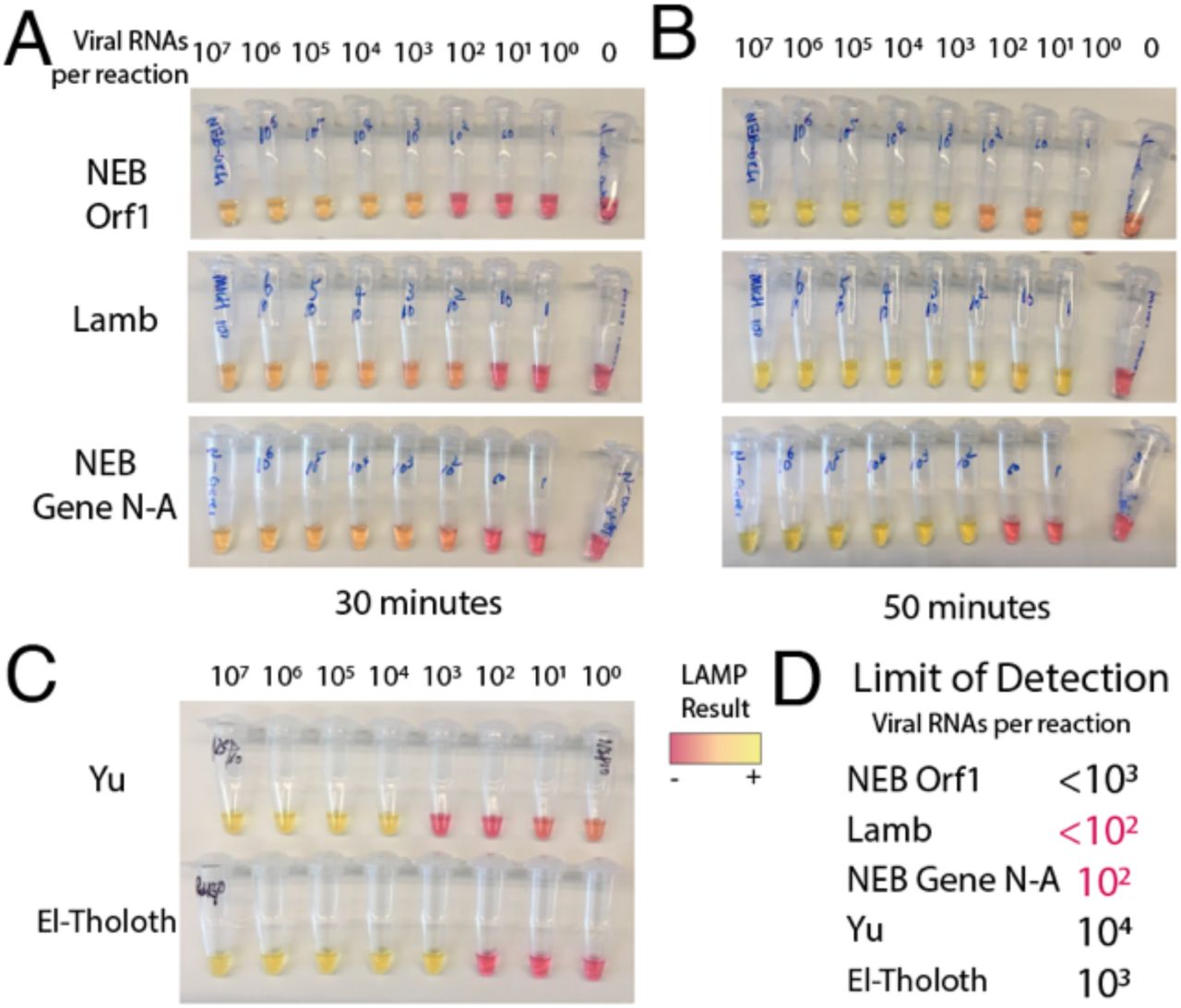
A) LAMP reactions produced colorimetric read-outs after 30 minutes for primer sets 1-3. Values indicate viral genome equivalent number of RNA per reaction. B) After 1 hour, some of the reactions became harder to interpret, as negative controls started turning yellow. C) Colorimetric LAMP assay for primer sets 4 and 5 after 1 hour. D) Approximate limits of detection (sensitivity to detect N number of viral genomes per reaction) were recorded, with primer sets 2 and 3 displaying the highest sensitivity, and no background amplification.
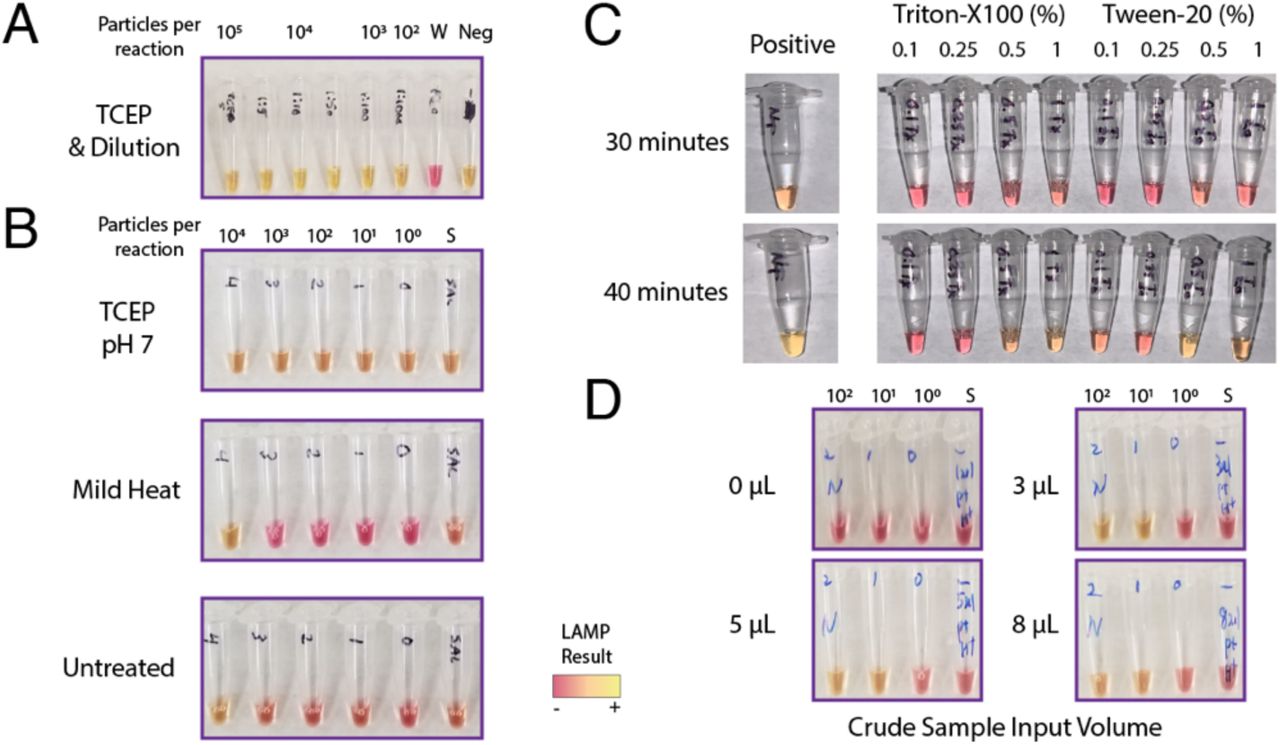
A) The HUDSON method saliva heat and chemical method was applied but TCEP pH caused false colorimetric shift. B) Neutral pH formulated TCEP still induced colorimetric shift without viral RNA. Mild heat was not sufficient to improve particle detection, as it matched untreated conditions. C) Low levels of detergents Triton-X and Tween-20 inhibited LAMP detection at even low levels at 30 minutes. D) The amount of crude saliva input to the LAMP reaction was varied. 3 uL was optimal to differentiate between positive and negative samples at 30 minutes. Increased volume did not increase the speed or sensitivity of the reactions. W = water. Neg = negative control saliva.

A) Colorimetric LAMP reactions using pairs of primer sets all increase detection sensitivity versus the single NEB Gene N-A primer set alone, with no increase in spurious amplification at 30 minutes. Neg = negative control saliva.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A) Stabilizing buffers and ribonuclease inhibitor RNAsecure are compatible with LAMP. Colorimetric LAMP reactions proceed with saliva diluted 1:1 in either water, phosphate-buffered saline (PBS), TE (Tris 10 mM, EDTA 0.1 mM), and diluted TCEP (2.5 mM TCEP, 0.5 mM EDTA). B) A 15-minute isothermal (64 °C) heat pre-treatment enables detection of 103 viral particles from saliva. Addition of RNAsecure improves sensitivity down to 102 viral particles. No additional benefit of diluted HUDSON reagents was seen compared to TE. C) Thermo-labile proteinase K treatment results in low sensitivity. While improved by RNAsecure, it performs worse than standard proteinase K with 98° inactivation, or mild heat treatments in (B). S = saliva control. D) Guanidium hydrochloride (40 mM) is compatible with the LAMP reaction. RNAsecure boosts the sensitivity of this additive. Primer multiplexing in this buffer may further boost sensitivity to 101 particles per reaction. All reactions performed with NEB Gene N-A primers, except in (D) which also included Lamb et al. primers as indicated.
Acknowledgments
The following reagent was deposited by the Centers for Disease Control and Prevention and obtained through BEI Resources, NIAID, NIH: SARS-Related Coronavirus 2, Isolate USA-WA1/2020, Heat Inactivated, NR-52286.
These studies were supported by donations to the WUSM COVID Research fund, the Department of Genetics and the McDonnell Genome Institute, and NIH grants P30 CA91842 and UL1 TR000448. We thank the Alvin J. Siteman Cancer Center at Washington University School of Medicine (WUSM) and Barnes-Jewish Hospital, the Institute of Clinical and Translational Sciences (ICTS), and the Tissue Procurement Core, which provided saliva samples. The Siteman Cancer Center is supported in part by an NCI Cancer Center Support Grant #P30 CA091842 and the ICTS is funded by NCATS Clinical and Translational Science Award (CTSA) program grant #UL1 TR002345.
References Cited



