
Abstract
Analysis of genetic sequence data from the pandemic SARS Coronavirus 2 can provide insights into epidemic origins, worldwide dispersal, and epidemiological history. With few exceptions, genomic epidemiological analysis has focused on geographically distributed data sets with few isolates in any given location. Here we report an analysis of 20 whole SARS-CoV 2 genomes from a single relatively small and geographically constrained outbreak in Weifang, People’s Republic of China. Using Bayesian model-based phylodynamic methods, we estimate the reproduction number for the outbreak to be 2.6 (95% CI:1.5-5). We further estimate the number of infections through time and compare these estimates to confirmed diagnoses by the Weifang Centers for Disease Control. We find that these estimates are consistent with reported cases and there is unlikely to be a large undiagnosed burden of infection over the period we studied.
Introduction
We report a genomic epidemiological analysis of one of the first geographically concentrated community transmission samples of SARS-CoV 2 genetic sequences collected outside of the initial outbreak in Wuhan, China. These data comprise 20 whole genome sequences from confirmed COVID19 infections in Weifang, Shandong Province, People’s Republic of China. The data were collected over the course of several weeks up to February 10, 2020 and overlap with a period of intensifying public health and social distancing measures. Phylodynamic analysis allows us to evaluate epidemiological trends after seeding events which took place in mid to late January, 2020. The objective of our analysis is to evaluate epidemiological trends based on national surveillance and response efforts by Weifang Centers for Disease Control (CDC). This analysis provides an estimate of the initial rate of spread and reproduction number in Weifang City. In contrast to the early spread of COVID19 in Hubei Province of China, most community transmissions within Weifang took place after public health interventions and social distancing measures were put in place. We therefore hypothesize that genetic data should reflect a lower growth rate and reproduction number than was observed in Wuhan. A secondary aim is to estimate the total numbers infected and to evaluate the possibility that there is a large unmeasured burden of infection due to imperfect case ascertainment and a large proportion of infections with mild or asymptomatic illness.
To analyze the Weifang sequences, we have adapted model-based phylodynamic methods which were previously used to estimate growth rates and reproduction numbers using sequence data from Wuhan and exported international cases(Volz et al., 2020). This analysis has several constraints and requirements:
Importation of lineages from Wuhan
The outbreak in Weifang was seeded by multiple lineages imported at various times from the rest of China. We use a phylodynamic model that accounts for location of sampling. Migration is modeled as a bi-directional process with rates proportional to epidemic size in Weifang. The larger international reservoir of COVID19 cases serves as a source of new infections and is assumed to be growing exponentially over this period of time.
Nonlinear epidemiological dynamics in Weifang
The maximum number of daily confirmed COVID19 cases occurred on February 5, but it is unknown when the maximum prevalence of infection occurred. We use a susceptible-exposed-infectious-recovered (SEIR) model(Keeling and Rohani, 2011) for epidemic dynamics in Weifang. The model accounts for a realistic distribution of generation times and can potentially capture a nonlinear decrease in cases following epidemic peak.
Variance in transmission rates
(Lloyd-Smith et al., 2005).To estimate total numbers infected, the phylodynamic model must account for epidemiological variables which are known to significantly influence genetic diversity. Foremost among these is the variance in offspring distribution (number of transmissions per primary case). We draw on previous evidence based on the previous SARS epidemic which indicates that the offspring distribution is highly over-dispersed. High variance of transmission rates will reduce genetic diversity of a sample and failure to account for this factor will lead to highly biased estimates of epidemic size(Li et al., 2017). Recent analyses of sequence data drawn primarily from Wuhan has found that high over-dispersion was required for estimated cases to be consistent with the epidemiological record(Volz et al., 2020). Models assuming low variance in transmission rates between people would generate estimates of cases that are lower than the known number of confirmed cases. Separately, Grantz et al.(Grantz et al., ????) have found that high over-dispersion is required to reconcile estimated reproduction numbers with the observed frequency of international outbreaks. In this study, we elaborate the SEIR model to include a compartment(J) with higher transmission rates. The variance of the implied offspring distribution is calibrated to give similar overdispersion from the SARS epidemic.
Results
Despite an initial rapid increase in confirmed cases in Weifang in late January and early February, the number of confirmed cases by Weifang CDC show that outbreak peaked quite early and maximum number of cases ocurred on February 5. Phylodynamic analysis supports the interpretation that control efforts reduced epidemic growth rates and contributed to eventual control. Figure 1A illustrates the phylodynamic model which was co-estimated with the phylogeny which provides estimates of epidemiological parameters summarized in Table 1. Figure 1B shows the estimated time scaled phylogeny (maximum clade credibility) including 20 lineages sampled from distinct patients in Weifang and 33 genomes sampled from Wuhan and internationally.
Summary of primary epidemiological and evolutionary parameters, including Bayesian prior distributions and estimated posteriors. Posterior uncertainty is summerized using a 95% highest posterior density interval
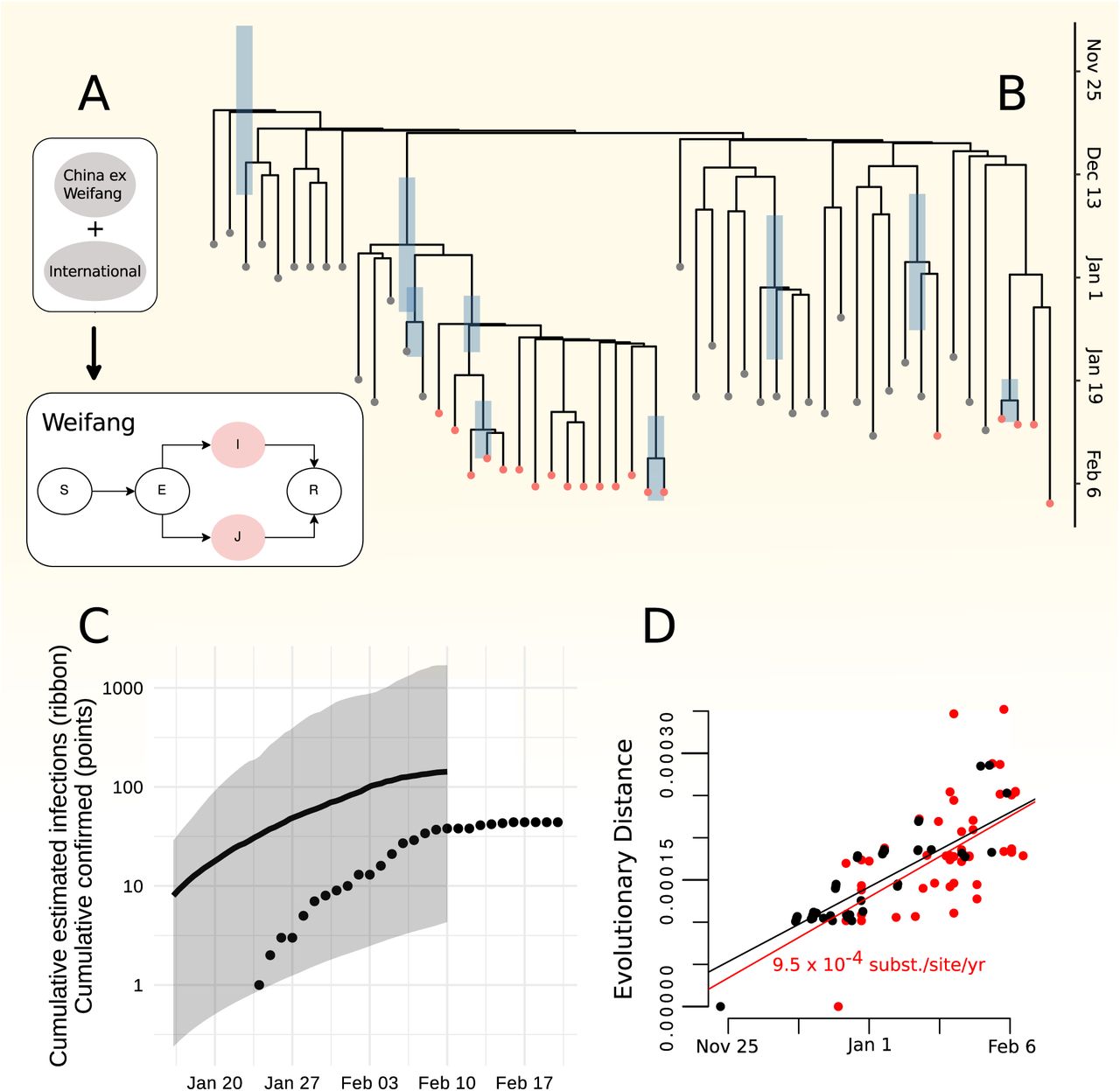
Phylodynamic estimates and epidemiological model. A. Diagram representing the structure of the epidemiological SEIR model which was fitted in tandem with the time scaled phylogeny. Colours correspond to the state of individuals sampled and represented in the tree (B). Note that infected and infectious individuals may occupy a low transmission state (I) or a high transmission rate state (J) to account for high dispersion of the reproduction number. B. A time scaled phylogeny co-estimated with epidemiological parameters. The colour of tips corresponds to location sampling. Red tips were sampled from Weifang, China. The credible interval of TMRCA is shown as a blue bar for all nodes with more than 50% posterior support. C. Cumulative estimated infections through time produced by fitting the SEIR model and the cumulative confirmed cases (points) reported by Weifan CDC. The shaded region shows the 95% HPD and the line shows the posterior median. D. A root to tip regression showing approximately linear increase in diversity with time of sampling.
Figure 1–Figure supplement 1. Maximum likelihood time tree.
Figure 1–Figure supplement 2. Tree posterior density plot.
Figure 1–Figure supplement 3. Tree posterior density plot.
The estimated number of infections is shown Figure 1C. The time series of confirmed cases should lag the estimated number of infected because of delays from infection to appearance of symptoms and delays from symptoms to diagnosis. We also expect that an unknown proportion of infections will be missed by the surveillance system due very mild, subclinical, or asymptomatic infection. Our estimates do not support the hypothesis that there was a very large hidden burden of infection in Weifang over the period that the sequence data were sampled. Indeed, our central estimate for the number infected on 10th February is only 142 and the credible intervals cover the 44 cumulative confirmed cases at the end of February.
We do not have suZcient data to detect a large decrease in epidemic growth rates as the epidemic progressed. We estimate R0 = 2.6 (95% HPD:1.5-5). The growth rate in estimated infections remained positive but decreased substantially over the sampling period. These estimates correspond to growth during a period when Weifang was implementing a variety of public health interventions and contact tracing to limit epidemic spread. These interventions included public health messaging, establishing phone hotlines, encouraging home isolation for recent vitors from Wuhan (January 23-26), optimizing triage of suspected cases in hospitals (January 24), travel restrictions(January 26), extending school closures, and establishing ‘fever clinics’ for consulatation and diagnosis(January 27)(Mao, 2020).
As well as providing novel epidemiological estimates, our results point to the significance of realistic modeling for fidelity of phylogenetic inference. The use of a model-based structured coalescent prior had large influence over estimated molecular clock rates and inferred TMRCAs. Figure Supplement 1 shows that maximum likelihood inference of time-scaled phylogenies produces a distribution of TMRCAs which are substantially different than the Bayesian model-based analysis. Choice of population genetic prior will have a large influence on phylogenetic inference based on sparse or poorly informative genetic sequence data. Among the 20 Weifang sequences included in this analysis, there is mean pairwise difference of only three single nucleotide polymorphisms and only approximately twice as much diversity observed among the remainder of the sequences we studied. There is correspondingly low confidence in tree topology (Figure Supplement 2), and only three clades had greater than 50% posterior support including one clade which had a monophyletic compoisition of 13 Weifang lineages. The earliest Weifang sequence was sampled on January 25 from a patient who showed first symptoms on January 16. These dates cover a similar range as the posterior TMRCA of all Weifang sequences (Figure Supplement 3).

A time scaled phylogeny estimated using IQTree and treedater and using the same data as used for the Bayesian analysis.

A tree density plot based on the posterior distribution of trees computed in BEAST2.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
The estimated posterior TMRCA among all Weifang lineages.
Discussion
Our analysis of 20 SARS-CoV 2 genomes from Weifang, China has confirmed independent observations regarding the rate of spread and burden of infection in the city. Surveillance of COVID19 is rendered difficult by high proportions of illness with mild severity and an unknown proportion of asymptomatic infection(Guan et al., 2020). The extent of under-reporting and case ascertainment rates has been widely debated. Analysis of genetic sequence data provides an alternative source of information about epidemic size which can be more robust to imperfect case ascertainment. We do not find evidence for a very large hidden burden of infection within Weifang. The relatively low estimate of R0 is consistent with a slower rate of spread outside of Wuhan and effective control strategies implemented in late January.
While the value of pathogen genomic analyses is widely recognized for estimating dates of emergence(Verity Hill, 2020; Gire et al., 2014) and identifying animal reservoirs(Zhou et al., 2020; Dudas et al., 2018), analysis of pathogen sequences also has potential to inform epidemic surveillance and intervention efforts. With few exceptions (Stadler, 2020; Bedford, 2020), this potential is currently not being realized for the international response to COVID19. It is worth noting that the analysis described in this report was accomplished in approximately 48 hours and drew on previously developed models and packages for BEAST2(Bouckaert et al., 2019; Volz and Siveroni, 2018). It is therefore feasible for phylodynamic analysis to provide a rapid supplement to epidemiological surveillance, however this requires rapid sequencing and timely sharing of data as well as randomized concentrated sampling of the epidemic within localities such as individual cities.
Methods and Materials
Epidemiological investigation, sampling, and genetic sequencing
As of 10 February 2020, 136 suspected cases, and 214 close contacts were diagnosed by Weifang Center for Disease Control and Prevention. 28 cases were detected positive with SARS-CoV-2. Viral RNA was extracted using Maxwell 16 Viral Total Nucleic Acid Purification Kit (Promega AS1150) by magnetic bead method and RNeasy Mini Kit (QIAGEN 74104) by column method. RT-qPCR was carried out using 2019 novel coronavirus nucleic acid detection kit (BioGerm, Shanghai, China) to confirm the presence of SARS-CoV-2 viral RNA with cycle threshold (Ct) values range from 17 to 37, targeting the high conservative region (ORF1ab/N gene) in SARS-CoV-2 genome. Metagenomic sequencing: The concentration of RNA samples was measurement by Qubit RNA HS Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA). DNase was used to remove host DNA. The remaining RNA was used to construct the single-stranded circular DNA library with MGIEasy RNA Library preparation reagent set (MGI, Shenzhen, China). Purified RNA was then fragmented. Using these short fragments as templates, random hexamers were used to synthesize the first-strand cDNA, followed by the second strand synthesis. Using the short double-strand DNA, a DNA library was constructed through end repair, adaptor ligation, and PCR amplification. PCR products were transformed into a single strand circular DNA library through DNA-denaturation and circularization. DNA nanoballs (DNBs) were generated with the single-stranded circular DNA library by rolling circle replication (RCR). The DNBs were loaded into the 2ow cell and pair-end 100bp sequencing on the DNBSEQ-T7 platform 8 (MGI, Shenzhen, China). 20 genomes were assembled with length from 26,840 to 29,882 nucleotides. The median age of patients was 36 (range:6-75). Two of twenty patients suffered severe or critical illness. Weifang sequences were combined with a diverse selection of sequences from China outside of Weifang and other countries provided by GISAIDElbe and Buckland-Merrett (2017). The new Weifang sequences are deposited in GISAID (EPI_ISL_413691, EPI_ISL_413692, EPI_ISL_413693, EPI_ISL_413694, EPI_ISL_413695, EPI_ISL_413696, EPI_ISL_413697, EPI_ISL_413711, EPI_ISL_413729, EPI_ISL_413746, EPI_ISL_413747, EPI_ISL_413748, EPI_ISL_413749, EPI_ISL_413750, EPI_ISL_413751, EPI_ISL_413752, EPI_ISL_413753, EPI_ISL_413761, EPI_ISL_413791, EPI_ISL_413809).
Mathematical model
The phylodynamic model is designed to account for nonlinear epidemic dynamics in Weifang, a realistic course of infection (incubation and infectious periods), migration of lineages in and out of Weifang, and variance in transmission rates which can influence epidemic size estimates. The model of epidemic dynamics within Weifang is based on a susceptible-exposed-infectious-recovered (SEIR) model. We elaborate the model with with an additional compartment J which has a higher transmission rate (r-fold higher) than the I compartment.Upon leaving the incubation period individuals progress to the J compartment with probability ph, or otherwise to I. The model is implemented as a system of ordinary differential equations:
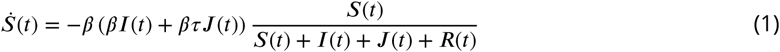
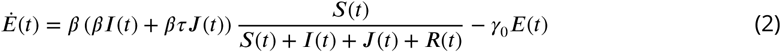



We also model an exponentially growing reservoir Y (t) for imported lineages in to Weifang. The equation governing this population is

Migration is modeled as a bidirectional process which only depends on the size of variables in the Weifang compartment and thus migration does not influence epidemic dynamics; it will only influence the inferred probability that a lineage resides within Weifang. For a compartment X (E,I, or J), ηis the per lineage rate of migration out of Weifang and the total rate of migration in and out of Weifang is ηX.
During phylodynamic model fitting β and p are estimated. Additionally, we estimate initial sizes of Y, E, and S. Other parameters are fixed based on prior information. We 1x 1/γ0 = 4.1days and 1/γ1 = 3.8 days. We set ph = 0.20 and τ = 74 which yields a dispersion of the reproduction number that matches a negative binomial distribution with k = 0.22 if R0 = 2, similar to values estimated for the 2003 SARS epidemic (Lloyd-Smith et al., 2005).
Phylogenetic analysis
We aligned the 20 Weifang sequences using MAFFT(Katoh and Standley, 2013) with a previous aligment of 35 SARS-CoV 2 sequences from outside of Weifang(Volz et al., 2020). Maximum likelihood analysis was carried using IQTree(Minh et al., 2019) with a HKY+G4 substitution model and a time-scaled tree was estimated using treedater 0.5.0(Volz and Frost, 2017). Two outliers according to the molecular clock model were identi1ed and removed using ‘treedater’ which was also used to compute the root to tip regression.
Bayesian phylogenetic analysis was carried out using BEAST 2.6.1(Bouckaert et al., 2019) using a HKY+G4 substitution model and a strict molecular clock. The phylodynamic model was implemented using the PhyDyn package(Volz and Siveroni, 2018) using the QL likelihood approximation and the RK ODE solver. The model was fitted by running 8 MCMC chains in parallel and combining chains after removing 50% burn-in.
The ggtree package was used for all phylogeny visualizations(Yu et al., 2017). Code to replicate this analysis and and BEAST XML 1les can be found at https://github.com/emvolz/weifang-sarscov2.
Funding
This work was supported by Centre funding from the UK Medical Research Council under a concordat with the UK Department for International Development. NIHR. J-IDEA. This work was also supported by a grant from the Special Project for Prevention and Control of Pneumonia of New Coronavirus Infection in Weifang Science and Technology Development Plan in 2020 (2020YQFK015) to Associate Senior Technologist Qing Nie. Role of the Funders: All funders of the study had no role in study design, data analysis, data interpretation, or writing of the report.
Acknowledgements
We gratefully acknowledge China National GeneBank at Shenzhen, China for the sequencing strategy and capacity support. We also gratefully acknowledge the laboratories that have contributed publicly available genomes via GISAID: Shanghai Public Health Clinical Center & School of Public Health, Fudan University, Shanghai, China, at the National Institute for Viral Disease Control and Prevention, China CDC, Beijing, China, at the Institute of Pathogen Biology, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, China, at the Wuhan Institute of Virology, Chinese Academy of Sciences, Wuhan, China, at the Department of Microbiology, Zhejiang Provincial Center for Disease Control and Prevention, Hangzhou, China, at the Guangdong Provincial Center for Diseases Control and Prevention at the Department of Medical Sciences, at the Shenzhen Key Laboratory of Pathogen and Immunity, Shenzhen, China, at the Hangzhou Center for Disease and Control Microbiology Lab, Zhejiang, China, at the National Institute of Health, Nonthaburi, Thailand, at the National Institute of Infectious Diseases, Tokyo, Japan, at the Korea Centers for Disease Control & Prevention, Cheongju, Korea, at the National Public Health Laboratory, Singapore, at the US Centers for Disease Control and Prevention, Atlanta, USA, at the Institut Pasteur, Paris, France, at the Respiratory Virus Unit, Microbiology Services Colindale, Public Health England, and at the Department of Virology, University of Helsinki and Helsinki University Hospital, Helsinki, Finland, and at the University of Melbourne, Peter Doherty Institute for Infection and Immunity, Melbourne, Australia, at the Victorian Infectious Disease Reference Laboratory, Melbourne, Australia, at the Public Health Virology Laboratory, Brisbane, Australia and at the Institute of Clinical Pathology and Medical Research, University of Sydney, Westmead, Australia.



