
Abstract
Background Autism Spectrum Disorder (ASD) is a neurodevelopmental disorder characterized by persistent deficits in social communication and interaction, along with restricted and repetitive behaviour patterns, interests or activities. Its prevalence has risen over the past few years, being four times more common in boys than girls. The cause of ASD is unclear, its etiology involves genetic, environmental, and gene-environment interactions. While past studies highlighted clinical genetic risks, genetic complexity of ASD, with variants of diverse frequencies, type, and inheritance patterns, requires further exploration for better management of disease. Researches have shown that the whole exome sequencing can be used to identify genetic variants associated with genetically heterogeneous conditions. The purpose of this study is to identify genetic variants by employing whole exome sequencing in an Indian ASD patient.
Methods A female patient of age within 0-5 years, having characteristic features like hyperactivity and language impairment, was investigated and diagnosed using DSM-5 criteria. Peripheral blood sample collection was done followed by DNA extraction and whole exome sequencing. Variants analysis, identification and annotation were done using bioinformatics tools and databases. Identified pathogenic variants were reconfirmed by Sanger sequencing.
Results and conclusion Our study uncover four genetic variations, comprising three missense variations in KIF1A (c.3839C>T), SETD5 (c.314A>C), MAPK81P3 (c.2849C>T), and one-stop gain variation in ERMARD (c.1523G>A). The ERMARD stop gain variation, predicted to induce nonsense-mediated decay, alter normal protein function through truncation and classified as likely pathogenic based on the ACMG guidelines and current available scientific evidence. In conclusion, we identified a likely pathogenic variant in ERMARD along with three missense variants in KIF1A, SETD5 and MAPK81P3 respectively. These findings suggest the potential contribution of ERMARD mutations to ASD susceptibility, emphasizing the need for further validation through functional studies.
Introduction
Autism Spectrum Disorder (ASD) is a common heterogenous lifelong neurodevelopmental condition ranging from mild to severe and characterized by persistent deficits in social communication and social interaction, restricted, repetitive patterns of behaviour, interests, or activities with unusual sensory-motor functions [1, 2]. Due to the absence of reliable biomarkers, the diagnosis most often is based upon the behaviour of the child. In recent years, its prevalence has gradually increased and become four times more common among boys than girls [3]. The estimated global prevalence of ASD is one in 100 [4]. The reported prevalence of ASD in South Asia is estimated to be one in 93 [5]. In India, the estimated prevalence of ASD in rural areas is 0.11% while in urban areas it is 0.09% (ages 1-18 years) [6]. ASD is clinically heterogeneous, some individuals presenting mild symptoms and others experiencing severe symptoms with a range of co-occurring physical and mental health conditions [7]. The etiology of ASD has not been understood, studies have shown that it may be multifactorial; genes, environment and gene-environment interactions play important role in the pathogenesis [8]. Large number of genes reported to be involved in the pathogenesis ASD, majority of which expressed in neuronal cells and enriched in maturing neurons [9,10,11,12] and thought to converge on common pathways affecting neuronal and synaptic homeostasis [13]. Pathway network analyses of gene ontologies suggest that, genes contributing to the core features of ASD may also contribute to other vulnerabilities, that is important molecular mechanisms leading to multiple systemic comorbidities that also overlap with other conditions [14]. Variations in multiple genes show the strong evidence of involvement of genetic factor in the pathogenesis of ASD [15]. Twin studies suggest the heritability of ASD to be 64%-91% [16]. Previous studies show the chromosomal abnormalities, copy number variations (CNVs) and single nucleotide variations (SNV) have been associated with ASD [17]. Rare or de novo genetic variants are identified in 5%-20% of individuals with ASD, and more often associated with complex medical presentation [18]. Rare variants causing ASD risk collectively encompass hundreds of genes [19], while copy-number variant and de novo protein-altering mutations show extreme locus heterogeneity [20]. In recent years development in genomic sequencing have transformed variant discovery, different approaches have been used to discover the genetic variants associated with ASD. Whole exome sequencing has been used to identify rare and novel genetic variation related to neurodevelopmental disorders [21] and have greatly improved the chance of identifying known as well as novel responsible genes [22, 23]. Studies has reported the combining clinical and molecular diagnosis is fundamental to deepen the knowledge of the pathogenic mechanisms of neurodevelopmental disorders underlying medical conditions and to develop personalized treatments [24].
In the present study, WES was performed for a patient sample with a diagnosis of an ASD related phenotype. We identified four genetic variations, including three missense and one stop gain variation. We select the variant which are predicted to alter normal protein function through protein truncation and classified as likely pathogenic for the reported phenotype based on current available scientific evidence using ACMG guideline [25].
In conclusion findings of this study provide valuable insights for pathogenicity of genetic variations and shed light on the underlying molecular process involved in ASD and confirm the efficacy of WES in detecting pathogenic variants in ASD candidate genes.
Material and Methods
Recruitment of patient and sample collection
Patient was enrolled from West Bengal, India. The study protocol was approved by the Institutional Ethics Committee of Centre for Genetic Disorders, Institute of Science, Banaras Hindu University, Varanasi.
The diagnosis of ASD was done according to the American Psychiatric Association’s Diagnostic and Statistical Manual of Mental Disorders (DSM-5) criteria [1] and ICD-10 [International Classification of Diseases, Tenth Revision] [26], and also evaluation was done using standard scale, IASQ (The Indian Autism Screening Questionnaire) [27].
As the proband was minor, peripheral blood sample was collected after obtaining the written informed consent from parents.
DNA Extraction and Whole exome sequencing
Genomic DNA was extracted using salting out method [28]. Whole exome sequencing was performed on genomic DNA sample of the proband. Sequencing of the protein coding regions of approximately 30 Mb of the human exome (targeting approximately 99% of the regions in CCDS and Refseq) was performed using Illumina next generation sequencing (NGS) systems at a mean depth of 50-60X and percentage of bases covered at 20X depth >90% in the target region.
Variant filtration and identification
Alignment of obtained sequences to human reference genome (GRCh37/hg19) was done using BWA-mem aligner. Variant calling was obtained using Genome Analysis ToolKit (GATK). Duplicate reads identification and removal, Base quality recalibration and re-alignment of reads based on indels were done using inbuilt Sentieon modules [29]. Sention’s Haplotype caller module was used to identify the variants which were relevant to the clinical indications along with the Deep variant analysis pipeline on Google cloud platform which was used as a secondary pipeline to call genetic variants [30]. Quality checks (QC) were performed on all VCF files to exclude variants where sequencing was of poor quality. Additional QC metrics includes total homozygous and heterozygous calls (SNVs and indels), proportion of variant calls that were common, number of variants falling into different annotated consequence categories, number of extreme heterozygous (alternative allele proportion 0.8).
Variant annotation and classification
The following public databases were used for annotation of identified variants: OMIM, GWAS, GNOMAD, 1000 Genomes database [31, 32, 33]. For the interpretation of variants, the American College of Medical Genetics and Genomics (ACMG) 2015 guidelines were used [25].
Sanger sequencing
To confirm the ERMARD variant, we performed Sanger sequencing of exon 16, including the flanking intron sequences of the gene (NM_018341.3) in the proband. PCR was done with specific primer pairs to amplify DNA, followed by purification of the PCR product. Subsequently, the purified PCR product underwent Sanger sequencing using an ABI 3500 Genetic Analyzer (Applied Biosystems, USA) according to the manufacturer’s protocol. The Sanger sequencing results were analysed with sequence scanner software 2 v2.0. Visualization of DNA sequences were performed by Finch TV v1.4.0 (Geospiza, PerkinElmer, USA) software.
Results
Clinical Description
This study encompasses a single Indian family, with the proband being a female of age within 0-5 years. She exhibited severe hyperactivity, speech regression, and an inability to articulate meaningful words.
She has no family history of ASD or other neurodevelopmental disorders.
WES Analysis
Whole exome sequencing data revealed a likely pathogenic stop-gain variant c.1523G>A in exon 16 of the ERMARD gene on chromosome 6 along with three missense variants; KIF1A (c.3839C>T), SETD5 (c.314A>C), MAPK81P3 (c.2849C>T), classified as variant of uncertain significance based on current available scientific evidence for the reported phenotype [Table 1].
Sanger sequencing confirmed the heterozygous ERMARD c.1523G>A variant in the proband [Figure 1]. This stop gain variant has not been previously reported and it is not present in gnomAD and 1000 genomes databases. This variation occurs upstream in exon 16 of ERMARD is predicted to be a nonsense mediated decay which alter normal protein function through protein truncation.

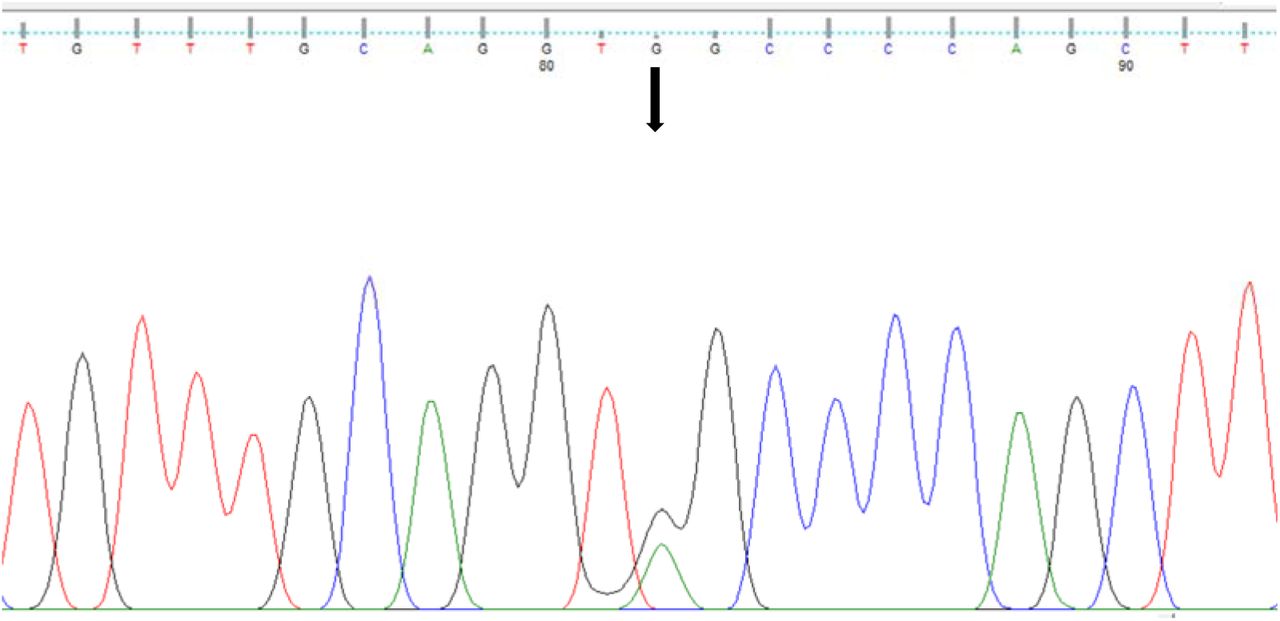{kind=link}
Confirmation of heterozygous variant c.1523G>A (denoted by arrow) by sanger sequencing.
Discussion
In present study, we identified a likely pathogenic variant c.1523G>A of ERMARD gene along with three variants of uncertain significance in genes; KIF1A (c.3839C>T), SETD5 (c.314A>C) and MAPK8IP3 (c.2849C>T) for the reported phenotype of a patient having characteristic features of ASD. These variations have not been reported previously as a pathogenic or benign and also not present in gnomAD and 1000 genomes databases.
ERMARD (ER membrane associated RNA degradation) gene encodes a protein with two transmembrane domains near the C-terminus and localised in the endoplasmic reticulum. Also known as C6ORF70, it is present on chromosome 6q27 [34]. According to previous studies the heterozygous mutations in the ERMARD have been associated with Periventricular nodular heterotopia-6 (PVNH6), a disease characterized by delayed psychomotor development, delayed speech, strabismus, onset of seizures with hypsarrhythmia and brain MRI showing bilateral periventricular nodular heterotopia in the frontal horns [34].
KIF1A encodes a motor protein that is involved in the anterograde transport of synaptic vesicle precursors along axons [35]. Mutations in KIF1A have been associated with a wide range of conditions including recessive mutations causing hereditary sensory neuropathy and hereditary spastic paraplegia [35,36] and de novo dominant mutations causing intellectual disability, cerebellar atrophy, spastic paraparesis, optic nerve atrophy, peripheral neuropathy, and epilepsy [37]. A de novo dominant missense variant has been reported in a patient presenting with ASD, spastic paraplegia and axonal neuropathy [38].
SETD5 is located on chromosome 3p25.3 and encodes the SETD5 protein composed of 1442 amino acids [39], and consists of 31 exons and is ubiquitously expressed in human tissues such as the brain, thyroid, skin, ovary, lung and endometrium [40, 41]. SETD5 contains a SET domain and is thus annotated as a candidate protein of lysine methyltransferase, which methylates H3K36 up to the tri-methyl form (H3K36me3) [40, 42, 43]. Autosomal dominant mental retardation-23 (MRD23) is caused by heterozygous mutation in the SETD5 gene, characterized by moderate to severe intellectual disability, delayed psychomotor development in infancy, poor speech development, obsessive-compulsive behaviour, hand-flapping and features of autism [44].
MAPK8IP3 encodes a member of the kinesin superfamily of proteins and plays a role in axonal transport [45]. Heterozygous mutation in the MAPK8IP3 caused neurodevelopmental disorder with or without variable brain abnormalities (NEDBA) [46].
In conclusion, findings of this study suggests that mutation in ERMARD with other reported variation in genes: KIF1A, SETD5 and MAPK8IP3 may cause the reported phenotype. Further functional studies in cell and animal models are needed to elucidate the role of variant in the pathogenesis of ASD.
Data Availability
All data produced in the present work are contained in the manuscript
References




