
Abstract
Importance While systemic inflammation has been implicated in the aetiology of selected neurodegenerative disorders, its role in the development of amyotrophic lateral sclerosis (ALS) is untested.
Objective To quantify the relationship of C-reactive protein (CRP), an acute-phase reactant and marker of systemic inflammation, with ALS occurrence.
Design, Setting, Participants UK Biobank, a prospective cohort study of 502,649 participants who were aged 37 to 73 years when examined at research centres between 2006 and 2010.
Exposure Venous blood was collected at baseline in the full cohort and assayed for CRP. Repeat measurement was made 3-7 years later in a representative subgroup (N=14,514) enabling correction for regression dilution.
Main Outcome(s) and Measure(s) ALS as ascertained via national hospitalisation and mortality registries. We computed multi-variable hazard ratios with accompanying 95% confidence intervals for log-transformed CRP expressed as standard deviation and tertiles.
Results In an analytical sample of 400,884 individuals (218,203 women), a mean follow-up of 12 years gave rise to 231 hospitalisations and 223 deaths ascribed to ALS. After adjustment for covariates which included health behaviours, comorbidity, and socio-economic status, a one standard deviation higher log-CRP was associated with elevated rates of both ALS mortality (hazard ratios; 95% confidence intervals: 1.32; 1.13, 1.53) and hospitalisations (1.20; 1.00, 1.39). There was evidence of dose-response effects across tertiles of CRP for both outcomes (p for trend≤0.05). Correction for regression dilution led to a strengthening of the relationship with CRP for both mortality (1.62; 1.27, 2.08) and hospitalisations (1.37; 1.05, 1.76) ascribed to ALS.
Conclusions and Relevance Higher levels of CRP, a blood-based biomarker widely captured in clinical practice, were associated with a higher subsequent risk of ALS.
Question Is C-reactive protein (CRP), a marker of systemic inflammation widely used in clinical practice, associated with later risk of amyotrophic lateral sclerosis (ALS)?
Findings Following 11 years disease surveillance in 400,884 individuals (218,203 women), after adjustment for covariates and correction for regression dilution, a one standard deviation higher CRP levels were associations with both mortality (hazard ratio; 95% confidence interval: 1.62; 1.27, 2.08) and hospitalisations (1.37; 1.05, 1.76) ascribed to ALS.
Meaning In the present study, CRP has a dose-response relationship with the risk of later ALS.
Introduction
Amyotrophic lateral sclerosis (ALS), also known as motor neuron disease, involves the unabated degeneration of nerve cells responsible for voluntary muscle movement. With there being no effective treatment, death from respiratory failure typically occurs within 3 years of symptom emergence.1,2 This brings into sharp focus the need for primary prevention research, yet series of studies examining the role of modifiable risk factors, including biomarkers, have revealed disappointing results.3-6
ALS is characterised by neuro-inflammation7 and there is some evidence implicating C-reactive protein (CRP), an acute-phase reactant and widely used marker of systemic inflammation, in the disease process. In animal models, higher levels of CRP appear to increase the permeability of the blood–brain barrier, so triggering microglial activation8 leading to increased release of proinflammatory cytokines, neuroinflammation, and cell death in the brain.9 Evidence for a role of CRP in the aetiology of ALS in humans is, however, largely circumstantial. While case-control studies have found that people with established ALS had higher CRP levels relative to the disease-free,10,11 this could be a consequence of the condition rather than a cause. Relatedly, surveillance of ALS patients reveals those with elevated systemic inflammation experienced a greater subsequent burden of disease severity, disability, and case fatality.10,12,13
The absence of evidence from well-characterised prospective studies on the potential role of CRP as a risk indicator for the onset of ALS is in part ascribed to the rarity of this disorder, rendering datasets insufficiently powered to yield sufficient cases to facilitate analyses. In UK Biobank,14 a prospective cohort study, a pre-morbid measurement of CRP was made in a cohort of 0.5 million members of the general population. Importantly, the repeat capturing of this inflammatory marker provides an unusual opportunity to address the impact of well-documented time-dependent fluctuations in CRP when measurement on a single baseline occasion is likely to lead to an underestimation of the true magnitude of the relationship with ALS.15
Methods
UK Biobank is a UK-wide, on-going, closed, prospective cohort study. Described in detail elsewhere,5 between 2006 and 2010, 502,649 people aged 37 to 73 years attended 22 geographically disparate research clinics where they completed a questionnaire, underwent an interview, and took part in a medical examination. Ethical approval was obtained from the National Health Service National Research Ethics Service with all participants providing written consent. Using pseudonymised data, the present analyses did not require additional permissions. The present report follows STROBE guidelines for the presentation of original epidemiological research.16
Assessment of baseline characteristics
Ethnicity was self-reported and categorised as White, Asian, Black, Chinese, Mixed, or other ethnic group.17 Cigarette smoking and physical activity were measured using standard enquiries.18 Self-reported physician diagnosis was collected for ALS, vascular or heart problems, diabetes, and cancer, and study members were asked if they had ever been under the care of a psychiatrist.19 The Townsend deprivation index, widely-used as an indicator of neighbourhood socioeconomic circumstances,20 is continuously scored with higher values denoting greater deprivation.
Height and weight were measured directly and body mass index was calculated using the usual formulae (weight, kg/height2, m2).21 Forced expiratory volume in one second, a measure of pulmonary function, was quantified using spirometry with the best of three technically satisfactory exhalations used in our analyses. The best result from 3 trials on each hand using a hydraulic hand dynamometer provided a measure of handgrip strength. Non-fasting venous blood was drawn, with assaying conducted at a dedicated central laboratory for CRP, glycated haemoglobin (HbA1c), and high-density lipoprotein (LDL) cholesterol.22 At resurvey, 3-7 years after baseline examination, a random sample of baseline study members (N=17,835) had their CRP levels reassessed.
Ascertainment of amyotrophic lateral sclerosis during follow-up
Study participants were linked to the National Health Service’s (NHS) Central Registry which provided vital status data on study members and, where applicable, cause of death.23 Linkage was also made to hospital in-patients records via the NHS Hospital Episode Statistics, a registry of all hospitalisations in the UK.24 Using both databases, ALS was denoted by the World Health Organization International Classification of Disease (version 10) code G122.
Statistical analyses
In all analyses, to address concerns regarding reverse causality – the notion that ALS might influence CRP25 rather than the opposite – we excluded 85 people who self-reported ALS at baseline medical examination and/or were hospitalised with the disease before study induction. Additionally, to capture study members with potentially subclinical (undiagnosed) ALS, we left-censored study members such that those who were hospitalised for, or died from, the condition within the first 3 years of baseline were also excluded (N=61). Lastly, study members with a CRP value ≥10.0, indicative of an acute infection, were also excluded.26 This resulted in analytical sample of 400,884 (218,203 women) with full data on CRP, covariates, and ALS outcomes.
To summarise the association between CRP and ALS, we used Cox proportional hazards regression to compute hazard ratios with accompanying 95% confidence intervals.27 In these analyses, calendar period was the time scale and study members were censored at date of hospitalisation or death from ALS, or end of follow-up (23 March 2021 for mortality, 5 May 2021 for hospitalisation) – whichever came first. CRP was utilised as a standardised, log-transformed linear term (mean=0, SD=1) and as tertiles. To examine potential non-linearity in the association between CRP levels and rates of ALS, we included CRP in models using natural cubic splines. To account for regression dilution bias, we also ran models including a dilution corrected value for CRP following a univariate regression calibration procedure.15 This involved regressing the second measure on the first and using the model predictions to calculate corrected CRP values for the full analytic sample. Using this derived value for CRP in the Cox model, confidence intervals were estimated via bootstrapping (1,000 replications).
Results
In table 1 we show baseline data according to tertiles of CRP. As anticipated, less favourable risk factor levels were universally apparent in people with higher CRP. For instance, people with higher levels of CRP tended to be older, have higher body mass index, lower lung function, and lower socio-economic position. Study members with higher CRP also had a greater prevalence of cigarette smoking, physical inactivity, and an array of morbidities, including vascular disease, cancer, diabetes, and mental illness.
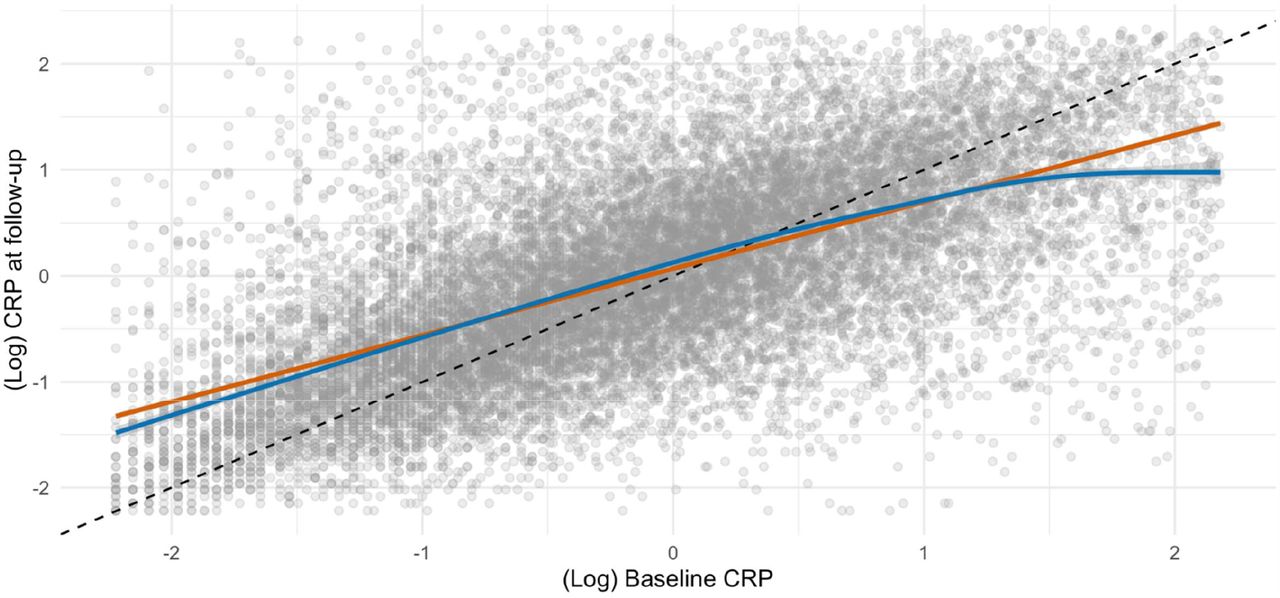
Mortality surveillance resulted in 223 deaths attributed to ALS, and in these analyses higher baseline CRP levels were related to a higher subsequent risk of this disorder (table 2 and supplemental figure 1). Thus, after adjustment for multiple covariates, the highest tertile of systemic inflammation was associated with a 75% increase in the risk of death from ALS (hazard ratio; 95% confidence interval: 1.75; 1.21, 2.53). A dose-response relationship was also apparent (p-value for trend <0.001) such that the central tertile of CRP had intermediate risk (1.46; 1.02, 2.08). When expressed per standard deviation increase in log of baseline CRP, there was a 32% increase in the risk of ALS (1.32; 1.13, 1.53). When we utilised resurvey data on CRP levels to examine the impact of repression dilution (correlation coefficient for survey-resurvey CRP was 0.64 [N= 14,514, p-value < 0.001; figure 1]), the magnitude of relative risk per SD approximately doubled with this correction (1.62; 1.27, 2.08).

A mean follow-up of 12.0 years (range 0.01-14.4) gave rise to 231 hospitalisations for ALS (117 women). The pattern of association for hospitalisation and CRP was similar to that seen for mortality but effect estimates were typically of lower magnitude and did not always achieve statistical significance at conventional levels (table 2 and supplemental figure 1). Again, a strengthening of the CRP–ALS association was apparent after taking into account regression dilution bias. Thus, the multiply-adjusted hazard ratio per SD increase in CRP increased from 1.20 (1.03, 1.39) to 1.37 (1.05, 1.76).
We also conducted some sensitivity analyses. First, splines allowed us to scrutinise these apparent dose-response effects for CRP in relation to ALS (figure 2) by searching for any inflection that would have been hidden by analyses of the categorisation of CRP. There was a suggestion that the steepest elevation in risk was apparent at lower levels of CRP with a less pronounced increase thereafter. The shape of the association was broadly similar for both deaths and hospitalisations. Second, just as there will be variation in CRP values over time, levels of the covariates in the present study are similarly time-dependent. Correcting for covariate biomarkers (HbA1c, HDL, FEV1 and grip strength), in addition to CRP, revealed very similar results to those apparent when correcting for CRP alone: hospitalisation for ALS (1.37; 1.05, 1.80) and mortality from ALS (1.64; 1.24, 2.10).

Discussion
Our main findings were that, in a population initially apparently free of ALS, higher baseline CRP was related to a subsequently elevated risk of developing this neurodegenerative disorder. This association was stronger for ALS mortality relative to hospitalisation; strengthened by adjustment for multiple confounding factors; and of markedly higher magnitude after correction for regression dilution. The two non-genetic factors most closely linked to the development of ALS – age and sex28 – were recapitulated here. Thus, people who were older (hazard ratio; 95% confidence interval per decade increase: 2.08; 1.72, 2.50) and male (1.16; 0.90, 1.50) experienced elevated rates of hospitalisation (corresponding results for death from ALS were 1.20; 0.92, 1.56; and 2.42; 1.98, 2.95). This gives us some confidence in the novel results for CRP.
Comparison with other studies
Elevated levels of systemic inflammation have been shown to be prospectively associated with higher rates of other neurodegenerative disorders such as dementia29 and Parkinson’s disease.30 With this study being, to our knowledge, the first to specifically examine the role of pre-morbid CRP as a potential risk factor for ALS occurrence, direct comparison of our results with other investigations is not possible. In a study most closely resembling our own, however, haptoglobin, a little-utilised marker of systemic inflammation, was not related to the development of ALS 15 years later.31 In vitro and animal work suggesting that the actions of the neuroinflammation-promoting enzyme, cyclooxygenase-2, are inhibited by non-steroidal anti-inflammatory drugs.32,33 In a related epidemiological study, however, investigators examining regular use of nonsteroidal anti-inflammatory medication prior to symptom onset found no relationship with ALS. 34,35
That the association between CRP and ALS was robust to the adjustment of potential confounding and meditating factors raises the suggestion of a direct effect. The observation that the steepest elevation in risk was apparent at lower levels of CRP with a less pronounced increase thereafter suggests the mechanism relates to low-grade systemic inflammation. One possibility is that even modestly raised of CRP increase the permeability of the blood–brain barrier, so triggering microglial activation8 leading to enhanced release of proinflammatory cytokines, neuroinflammation, and cell death in the brain.36 It was not possible to directly explore these potential explanations using the present data, however.
Study strengths and limitations
The strengths of the present study include its novelty; the use of recapture data on CRP to examine regression dilution bias; its scale, which facilitates the accumulation of a sufficiently high number of cases for analyses of a rare neurodegenerative disorder alongside left-censoring to take into account reverse causality; and the well-characterised nature of the study participants which facilitates adjustment for multiple confounding factors.
Inevitably, however, our work has its weaknesses. First, the present study sample comprises only the 5.5% of the target population.14 As has been demonstrated,37,38 the data material is therefore inappropriate for estimation of risk factor or disease prevalence and for event incidence, including for ALS. These observations do not, however, seem to influence reproducibility of the association of established risk factors for important health outcomes such as vascular disease, selected cancers, and suicide.38 We think the same reasoning can be applied to the present analyses for ALS. Second, we did not have data on other markers of systemic inflammation such as interleukin 6 with which to draw comparison with the present results for CRP. Lastly, the outcomes herein represent advanced ALS and it is unknown if CRP is related to ALS at earlier stages of disease progression.
In conclusion, in the present study, higher levels of pre-morbid CRP, an inflammatory marker commonly captured in clinical practice, appeared to be associated with higher risk of incident ALS. These results warrant further research examining the mechanisms linking systemic inflammation to ALS pathology.
Data Availability
Data from UK Biobank upon application (http://www.ukbiobank.ac.uk/). Part of the present research has been conducted using the UK Biobank Resource under Application 10279.
Supplemental figures


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Footnotes
Access to data: Data from UK Biobank upon application (http://www.ukbiobank.ac.uk/). Part of the present research has been conducted using the UK Biobank Resource under Application 10279.
Funding: GDB is supported by the US National Institute on Aging (1R56AG052519-01; 1R01AG052519-01A1) and the UK Medical Research Council (MR/P023444/1; MR/X003434/1), and MK by the Wellcome Trust (221854/Z/20/Z), the UK Medical Research Council (MR/S011676/1), the US National Institute on Aging (R01AG056477), and the Academy of Finland (350426). These funders provided no direct financial or material support for the work, and had no role in study design, data collection, data analysis, data interpretation, or report preparation.
Email: david.batty{at}ucl.ac.uk
m.kivimaki{at}ucl.ac.uk
philipp.frank.16{at}ucl.ac.uk
crg{at}mrc.soton.ac.uk
liam.wright{at}ucl.ac.uk
Typing error for co-author ('Frank' not 'FRank')
References



