
Abstract
The extent to which low‐frequency (minor allele frequency (MAF) between 1–5%) and rare (MAF ≤ 1%) variants contribute to complex traits and disease in the general population is mainly unknown. Bone mineral density (BMD) is highly heritable, a major predictor of osteoporotic fractures, and has been previously associated with common genetic variants1,2,3,4,5,6,7,8, as well as rare, population‐specific, coding variants9. Here we identify novel non‐coding genetic variants with large effects on BMD (ntotal = 53,236) and fracture (ntotal = 508,253) in individuals of European ancestry from the general population. Associations for BMD were derived from whole‐genome sequencing (n = 2,882 from UK10K (ref. 10); a population‐based genome sequencing consortium), whole‐exome sequencing (n = 3,549), deep imputation of genotyped samples using a combined UK10K/1000 Genomes reference panel (n = 26,534), and de novo replication genotyping (n = 20,271). We identified a low‐frequency non‐coding variant near a novel locus, EN1, with an effect size fourfold larger than the mean of previously reported common variants for lumbar spine BMD8 (rs11692564(T), MAF = 1.6%, replication effect size = +0.20 s.d., Pmeta = 2 × 10−14), which was also associated with a decreased risk of fracture (odds ratio = 0.85; P = 2 × 10−11; ncases = 98,742 and ncontrols = 409,511). Using an En1cre/flox mouse model, we observed that conditional loss of En1 results in low bone mass, probably as a consequence of high bone turnover. We also identified a novel low‐frequency non‐coding variant with large effects on BMD near WNT16 (rs148771817(T), MAF = 1.2%, replication effect size = +0.41 s.d., Pmeta = 1 × 10−11). In general, there was an excess of association signals arising from deleterious coding and conserved non‐coding variants. These findings provide evidence that low‐frequency non‐coding variants have large effects on BMD and fracture, thereby providing rationale for whole‐genome sequencing and improved imputation reference panels to study the genetic architecture of complex traits and disease in the general population.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others

References
Richards, J. B. et al. Bone mineral density, osteoporosis, and osteoporotic fractures: a genome‐wide association study. Lancet 371, 1505–1512 (2008)
Styrkarsdottir, U. et al. Multiple genetic loci for bone mineral density and fractures. N. Engl. J. Med. 358, 2355–2365 (2008)
Styrkarsdottir, U. et al. New sequence variants associated with bone mineral density. Nature Genet. 41, 15–17 (2009)
Rivadeneira, F. et al. Twenty bone‐mineral‐density loci identified by large‐scale meta‐analysis of genome‐wide association studies. Nature Genet. 41, 1199–1206 (2009)
Duncan, E. L. et al. Genome‐wide association study using extreme truncate selection identifies novel genes affecting bone mineral density and fracture risk. PLoS Genet. 7, e1001372 (2011)
Koller, D. L. et al. Genome‐wide association study of bone mineral density in premenopausal European‐American women and replication in African‐American women. J. Clin. Endocrinol. Metab. 95, 1802–1809 (2010)
Xiong, D.‐H. et al. Genome‐wide association and follow‐up replication studies identified ADAMTS18 and TGFBR3 as bone mass candidate genes in different ethnic groups. Am. J. Hum. Genet. 84, 388–398 (2009)
Estrada, K. et al. Genome‐wide meta‐analysis identifies 56 bone mineral density loci and reveals 14 loci associated with risk of fracture. Nature Genet. 44, 491–501 (2012)
Styrkarsdottir, U. et al. Nonsense mutation in the LGR4 gene is associated with several human diseases and other traits. Nature 497, 517–520 (2013)
The UK10K Consortium The UK10K project identifies rare variants in health and disease. Nature http://dx.doi.org/10.1038/nature14962 (this issue)
Hindorff, L. A. et al. Potential etiologic and functional implications of genome‐wide association loci for human diseases and traits. Proc. Natl Acad. Sci. USA 106, 9362–9367 (2009)
Kiezun, A. et al. Exome sequencing and the genetic basis of complex traits. Nature Genet. 44, 623–630 (2012)
Gudbjartsson, D. F. et al. Large‐scale whole‐genome sequencing of the Icelandic population. Nature Genet. 47, 435–452 (2015)
Sulem, P. et al. Identification of a large set of rare complete human knockouts. Nature Genet. 47, 448–444 (2015)
Abecasis, G. R. et al. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65 (2012)
Richards, J. B., Zheng, H.‐F. & Spector, T. D. Genetics of osteoporosis from genome‐wide association studies: advances and challenges. Nature Rev. Genet. 13, 576–588 (2012)
Huang, J. et al. Improved imputation of low‐frequency and rare variants using the UK10K haplotype reference panel. Nature Comm. 6, 8111 (2015)
Xu, C., Tachmazidou, I., Walter, K., Ciampi, A., Zeggini, E. & Greenwood, C. M. T. Estimating genome‐wide significance for whole‐genome sequencing studies. Genet. Epidemiol. 38, 281–290 (2014)
Dixon, J. R. et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380 (2012)
Loomis, C. A. et al. The mouse Engrailed‐1 gene and ventral limb patterning. Nature 382, 360–363 (1996)
Adamska, M., MacDonald, B. T., Sarmast, Z. H., Oliver, E. R. & Meisler, M. H. En1 and Wnt7a interact with Dkk1 during limb development in the mouse. Dev. Biol. 272, 134–144 (2004)
Deckelbaum, R. A., Majithia, A., Booker, T., Henderson, J. E. & Loomis, C. A. The homeoprotein engrailed 1 has pleiotropic functions in calvarial intramembranous bone formation and remodeling. Development 133, 63–74 (2006)
Matise, M. P. & Joyner, A. L. Expression patterns of developmental control genes in normal and Engrailed‐1 mutant mouse spinal cord reveal early diversity in developing interneurons. J. Neurosci. 17, 7805–7816 (1997)
Sgaier, S. K. et al. Genetic subdivision of the tectum and cerebellum into functionally related regions based on differential sensitivity to engrailed proteins. Development 134, 2325–2335 (2007)
Ackert‐Bicknell, C. L. et al. Mouse BMD quantitative trait loci show improved concordance with human genome‐wide association loci when recalculated on a new, common mouse genetic map. J. Bone Miner. Res. 25, 1808–1820 (2010)
Zheng, H.‐F. et al. WNT16 influences bone mineral density, cortical bone thickness, bone strength, and osteoporotic fracture risk. PLoS Genet. 8, e1002745 (2012)
Medina‐Gomez, C. et al. Meta‐analysis of genome‐wide scans for total body BMD in children and adults reveals allelic heterogeneity and age‐specific effects at the WNT16 locus. PLoS Genet. 8, e1002718 (2012)
Movérare‐Skrtic, S. et al. Osteoblast‐derived WNT16 represses osteoclastogenesis and prevents cortical bone fragility fractures. Nature Med. 20, 1279–1288 (2014)
Ladouceur, M., Dastani, Z., Aulchenko, Y. S., Greenwood, C. M. T. & Richards, J. B. The empirical power of rare variant association methods: results from Sanger sequencing in 1,998 individuals. PLoS Genet. 8, e1002496 (2012)
Tang, H. et al. A large‐scale screen for coding variants predisposing to psoriasis. Nature Genet. 46, 45–50 (2014)
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009)
Mägi, R. & Morris, A. P. GWAMA: software for genome‐wide association meta‐analysis. BMC Bioinformatics 11, 288 (2010)
Yang, J. et al. Conditional and joint multiple‐SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nature Genet. 44, 369–375 (2012)
Voorman, A. A., Brody, J. & Lumley, T. SkatMeta: an R package for meta‐analyzing region‐based tests of rare DNA variants. (http://cran.r-project.org/web/packages/skatMeta) (2013)
Wu, M. C. et al. Rare‐variant association testing for sequencing data with the sequence kernel association test. Am. J. Hum. Genet. 89, 82–93 (2011)
Davydov, E. V. et al. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLOS Comput. Biol. 6, e1001025 (2010)
Chang, C. C. et al. Second‐generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015)
McLaren, W. et al. Deriving the consequences of genomic variants with the Ensembl API and SNP effect predictor. Bioinformatics 26, 2069–2070 (2010)
Hanks, M., Wurst, W., Anson‐Cartwright, L., Auerbach, A. B. & Joyner, A. L. Rescue of the En‐1 mutant phenotype by replacement of En‐1 with En‐2. Science 269, 679–682 (1995)
Acknowledgements
Full acknowledgements are listed in the Supplementary Information.
Author information
Authors and Affiliations
Consortia
Contributions
Principal Investigators: A.H., A.J., A.U., A.X.‐A., B.L., C.A.‐B., Ch.C., C.L., Cl.C., C.L.D., C.M.v.D., C.O., D.S.E., D.Ga., D.Go., D.Gr., D.H., D.Ki., D.M., E.D., E.O., F.Ri., F.Ro., G.D.S., J.B.R., J.D., J.Re., J.Ri., J.‐T.K., J.Tu., K.A., L.A.C., L.L., L.P.G.M.d.G., M.B., M.M.F., N.S., N.T., N.v.d.V., N.vS., P.R., R.D., R.D.J., R.L.P., S.G.W., S.H.R., T.H., T.P., T.S., U.P.‐K.,V.G., X.N.,Y.‐H.H. Genotyping: AOGC Consortium, A.U., B.M., B.W., C.L., C.M.v.D., C.N., C.O., C.W., D.C., D.Gr., E.D., E.O., F.Ri., G.G., G.Tr., J.Er., J.Jv.M., J.Re., J.Ri., J.‐T.K., J.v.R., M.B., M.C.F., M.J., M.Z., N.A., N.G.‐G., N.S., N.T., P.Ar., P.D., P.R., R.K., S.H.R., S.M., S.R., U.P.‐K., X.N. and Y.‐H.H. Phenotyping: A.E., A.H., A.L., AOGC Consortium, A.P.H., A.U., A.X.‐A., B.M., C.G., C.K., C.L., C.L.D., C.M.v.D., C.O., D.Go., D.Ka., D.Ki., D.M., E.D., E.N., E.O., F.E.M., F.K., F.Ri., F.Ro., G.H., J.B., J.C., J.Ei., J.O., J.Re., J.Ri., J.‐T.K., J.To., K.E., K.S., K.T., L.O., L.R., L.V., M.B., M.C.F., M.M.F., M.C.Z., M.K., M.Z., N.A., N.S., N.T., O.L., O.S., R.L.P., S.G.W., S.G., S.H.R., S.K., T.N., T.S. and U.P.‐K. Functional experiments: A.J., A.R.‐D., B.Ge, C.A.‐B., C.H., C.L., C.L.D., C.O., C.U., D.Ga., D.P., E.G., H.Y.P.‐M., J.D., J.F., K.Ch., Ma.M., M.H., N.S., O.S., S.B., S.C., S.‐H.C., St.W., T.K., T.P., U.P.‐K., W.C. and X.J. Data analysis: A.E., A.K., A.S., A.V.S., B.M., C.A.‐B., Ch.C., C.‐H.C., C.K., C.L., C.L.D., C.M.‐G., C.M.T.G., C.O., C.T.L., C.W., D.S.E., D.M.E., D.C., D.Ka., D.M., D.P., E.D., E.G., E.N., F.G., F.Ri., G.H., G.Th., H.‐F.Z., J.B.R., J.D., J.Er., J.F., J.Ha., J.Hu., J.K., J.v.R., K.Ch., K.E., K.W., L.A.C., L.H., L.M., L.O., L.R., L.V., M.B., M.C., M.H., M.K., N.A., N.S., N.T., O.L., P.Au., P.D., P.L., R.L., S.B., S.C., S.G.W., S.K., U.P., U.P.‐K., V.F., W.‐C.C., Y.‐H.H., Y.M. and Y.Z. Meta‐analysis: H.‐F.Z., V.F. and Y.‐H.H. Lead analysts: H.‐F.Z. and V.F. Wrote first draft: J.B.R.
Corresponding author
Ethics declarations
Competing interests
Authors from deCODE Genetics are employees of deCODE Genetics. Authors from 23andMe are employees of 23andMe. Remaining authors declare no competing financial interests.
Additional information
Source code used in preparation of results is available at https://github.com/richardslab/gefos.seq/. BMD discovery meta‐analysis results are available from http://www.gefos.org. Information pertaining to UK10K can be obtained from http://www.uk10k.org.
Lists of participants and their affiliations appear in the Supplementary Information.
Lists of participants and their affiliations appear in the Supplementary Information.
Extended data figures and tables
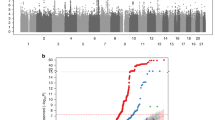
Extended Data Figure 1 Discovery single variant meta‐analysis.
a, Overall study design. b, From top to bottom, quantile–quantile plots for the sex‐combined single SNV meta‐analysis, sex‐stratified single SNV meta‐analysis (forearm phenotype consists solely of female‐only cohorts), and sex‐combined single SNV conditional meta‐analysis Plots depicts P values prior (blue) and after (red) conditional analysis on genome-wide significant variants (see Supplementary Methods). c, From top to bottom, Manhattan plots for sex‐combined meta‐analysis for lumbar spine BMD, femoral neck BMD, and forearm BMD. Each plot depicts variants from the UK10K/1000G reference panel with MAF > 0.5% across the 22 autosomes (odd, grey; even, black) against the −log10 P value from the meta‐analysis of 7 cohorts (dots). Also depicted are the subset variants from the reference panel that are also present in ref. 8 with P value <5 × 10−6 (diamonds). Variants with MAF < 5% and P < 1.2 × 10−6 are also depicted (red). d, Quantile–quantile plots for the sex‐combined meta‐analysis of lumbar spine, femoral neck, and forearm BMD for SNVs present across both exome‐sequenced and genome-sequenced and imputed cohorts, that is, SNV present only in genome-sequenced or imputed cohorts are not shown. e, Manhattan plot for the meta‐analysis of sex‐combined results for lumbar spine BMD for SNVs present in exome‐sequenced and genome-sequenced and imputed cohorts, that is, SNV present only in genome-sequenced or imputed cohorts are not shown (from left to right: lumbar spine, forearm and femoral neck BMD).
Extended Data Figure 2 Forest plots by cohort for genome‐wide significant loci from discovery meta‐analysis.
Forest plots for three BMD phenotypes are shown. Title of each plot includes gene overlapping the SNV and its genomic position on build hg19. P values are from fixed‐effect meta‐analysis (see Supplementary Information).
Extended Data Figure 3 Gene expression in human and mouse.
a, Quantification of Dock8 expression and its temporal pattern through RNA‐seq in cultured calvarial murine osteoblasts across day 2 through to day 18 of osteoblast development. Shown for comparison is Bglap, which encodes osteocalcin, a critical protein in osteoblasts. b, Quantification of expression of genome‐wide significant genes and their temporal pattern through RNA‐seq in cultured calvarial murine osteoblasts across day 2 through to day 18 of osteoblast development. c, Expression of EN1 mRNA in human cells presented as per cent of GAPDH mRNA. d, Expression of En1 in control and sdEn1 mice in purified osteoblast culture. For osteoblast marker gene expression, total mRNAs were purified from osteoblast cultures at day 10 and measured using quantitative real‐time PCR. mRNA levels were normalized relative to GAPDH mRNA. e. Real‐time PCR expression of control and sdEn1 as compared to 18S mRNA in whole vertebral bone extract. All data are shown as mean ± s.e.m. Significance computed by Student’s unpaired t‐test.
Extended Data Figure 4 Histological assessment of En1cre‐expressing cells in skeletal cells of the vertebra.
a, Lineage history of En1cre‐expressing cells in skeletal cells of the vertebra. The En1cre allele was combined with the R26LSL‐YFP reporter allele and examined using frozen fluorescent immunohistochemistry and alkaline phosphatase (AP) staining. Cell nuclei were detected with DAPI. YFP‐expressing cells have expressed Cre (En1) at some time in their history. In subpanel A, control animals lacking the R26LSL‐YFP reporter show low background YFP signal (green). In subpanel B, En1cre/+; R26LSL‐YFP/+ mice YFP‐expressing cells are detected in the growth plate chondrocytes of the vertebra (asterisk), trabecular bone lining cells (arrow) and osteocytes (arrowhead). Note, high fluorescent background staining in the marrow space. In subpanel C, the same section is shown stained for AP activity using the Fast Red substrate. Strong activity is present in the hypertrophic chondrocytes of the growth plate and trabecular bone lining cells (arrow). In subpanel D, alignment of the AP and YFP images shows that the trabecular lining cells co‐express AP and YFP. b, Co‐localization of En1 and alkaline phosphatase expression. Images of lumbar vertebrae sections (growth plate and trabecular bone regions, ×40 magnification) from two‐month old En1lacZ/+ mice (see Fig. 3b), stained for LacZ and alkaline phosphatase (AP), false‐coloured as indicated. Double‐positive cells are indicated by arrows, single‐positive cells are indicated by arrowheads (LacZ+) or asterisks (AP+). Except for some chondrocytes, most AP+ cells are also LacZ+, that is, express En1. The bone marrow was digitally removed, as it contains no AP+ cells.
Extended Data Figure 5 Micro‐CT results for control (En1flox/+) and self‐deleting En1 knockout (sdEn1, En1cre/flox) animals.
a, Trabecular bone micro‐CT images from lumbar vertebra 5. b, Morphological characteristics at lumbar vertebra 4, 5, and 6 (from bottom to top). c, d, Morphological characteristics of left femur trabecular bone (c) and left femur cortical bone (d). e, Micro‐CT parameter results for the comparison of control and sdEn1 animals at lumbar vertebra 5, femur trabecula, and femur cortical bone. Horizontal lines denote mean of observations. Significance between control and sdEn1 is calculated using an unpaired t‐test.
Extended Data Figure 6 Novel association from 7q31.3.
a, Chromatin interaction data from Hi‐C performed in H1 embryonic stem cells23 of a 2 Mb region encompassing rs148771817 (red and identified by arrow) and WNT16. b, The left axis denotes the association P value (red and green lines at P = 1.2 × 10−5 and 1.2 × 10−8, respectively). The novel genome‐wide significant SNV, rs148771817, within an intron of CPED1, and the lead genome‐wide significant SNV rs7776725 upstream to WNT16 (within FAM3C) are in low LD with each other. c, Allele frequency versus absolute effect size (in standard deviations) for forearm BMD of all previously identified genome‐wide significant variants (blue)8 and the novel variant within CPED1 (red), rs148771817 from replication meta‐analysis. The blue line denotes the mean of effect sizes for previously reported forearm BMD variants. d, Meta‐analysis summary statistics of rs148771817 conditioned on rs7776725.
Extended Data Figure 7 Regional plots of genome‐wide significant loci from single‐SNV association tests for forearm and femoral neck BMD.
Each regional plot depicts SNVs within 1 Mb of a locus’ lead SNV (x axis) and their associated meta‐analysis P value (−log10). SNVs are colour-coded according to r2 with the lead SNV (labelled, r2 calculated from UK10K whole‐genome sequencing data set). Recombination rate (blue line), and the position of genes, their exons and the direction of transcription are also displayed (below plot).
Extended Data Figure 8 Regional plots of genome‐wide significant loci from single‐SNV association tests from lumbar spine BMD.
Each regional plot depicts SNVs within 1 Mb of a locus’ lead SNV (x axis) and their associated meta‐analysis P value (−log10). SNVs are colour coded according to r2 with the lead SNV (labelled, r2 calculated from UK10K whole genome sequencing data set). Recombination rate (blue line), and the position of genes, their exons and the direction of transcription are also displayed (below plot).
Extended Data Figure 9 Region‐based association tests using skatMeta for windows of 30 SNVs and window step of 20 SNVs.
a, Left, quantile–quantile plots for forearm (FA) BMD, femoral neck (FN) BMD, and lumbar spine (LS) BMD. For each MAF range considered (<5% or < 1%), analysis was conducted across all variants, variant overlapping coding exons, and variants with GERP++ score >1. b, Right, Manhattan plots forearm BMD, femoral neck BMD, and lumbar spine BMD. For each MAF range considered (<5% or < 1%), analysis was conducted across all variants, variant overlapping coding exons, and variants with GERP++ score >1. Blue lines indicate genome‐wide suggestive (P = 1.2 × 10−6) thresholds and red lines indicate genome‐wide significant (P = 1.2 × 10−8) thresholds.
Extended Data Figure 10 Single variant analysis of signals from region‐based tests.
a, Drop‐one SNV (left) and drop‐one cohort (right) for genome‐wide significant 30 SNV windows for femoral neck and forearm BMD from skatMeta analysis. On left, for a given 30 SNV window, the −log10P of skatMeta test for 29 SNVs, excluding (that is, dropping) the SNV at position labelled on the x axis. On right, for given 30 SNV window on left, the −log10P of skatMeta test for all cohorts, excluding (that is, dropping) cohort labelled on x axis. b, Regional view of CPED1/WNT16 locus for forearm BMD. Significant SNVs from single variant meta‐analysis (rs148771817 and rs79162867, in blue) overlap significant regions found using region‐based test (red bars).
Supplementary information
Supplementary Information
This file contains Supplementary Text and Supplementary References. (PDF 1093 kb)
Supplementary Tables
This file contains Supplementary Tables 1-19. (XLSX 757 kb)
Rights and permissions
About this article

Cite this article
Zheng, H., Forgetta, V., Hsu, Y. et al. Whole‐genome sequencing identifies EN1 as a determinant of bone density and fracture. Nature 526, 112–117 (2015). https://doi.org/10.1038/nature14878
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature14878
This article is cited by
-
Genetic evidence of the causal relationship between chronic liver diseases and musculoskeletal disorders
Journal of Translational Medicine (2024)
-
Causal relationship between glycemic traits and bone mineral density in different age groups and skeletal sites: a Mendelian randomization analysis
Journal of Bone and Mineral Metabolism (2024)
-
Investigating the association between serum ADAM/ADAMTS levels and bone mineral density by mendelian randomization study
BMC Genomics (2023)
-
Osteoporosis and osteoarthritis: a bi-directional Mendelian randomization study
Arthritis Research & Therapy (2023)
-
Causal association of epigenetic aging and osteoporosis: a bidirectional Mendelian randomization study
BMC Medical Genomics (2023)
