
ABSTRACT
Previous studies have reported that a third dose of the BNT162b2 (Pfizer) COVID-19 vaccine increased antibody titers and protective efficacy. Here we compare humoral and cellular immune responses in 65 individuals who were vaccinated with the BNT162b2 vaccine and were boosted after at least 6 months with either Ad26.COV2.S (Johnson & Johnson; N=41) or BNT162b2 (Pfizer; N=24).
Previous studies have reported that a third dose of the BNT162b2 (Pfizer) COVID-19 vaccine increased antibody titers and protective efficacy1,2. Here we compare humoral and cellular immune responses in 65 individuals who were vaccinated with the BNT162b2 vaccine and were boosted after at least 6 months with either Ad26.COV2.S3 (Johnson & Johnson; N=41) or BNT162b24 (Pfizer; N=24) (Table S1).
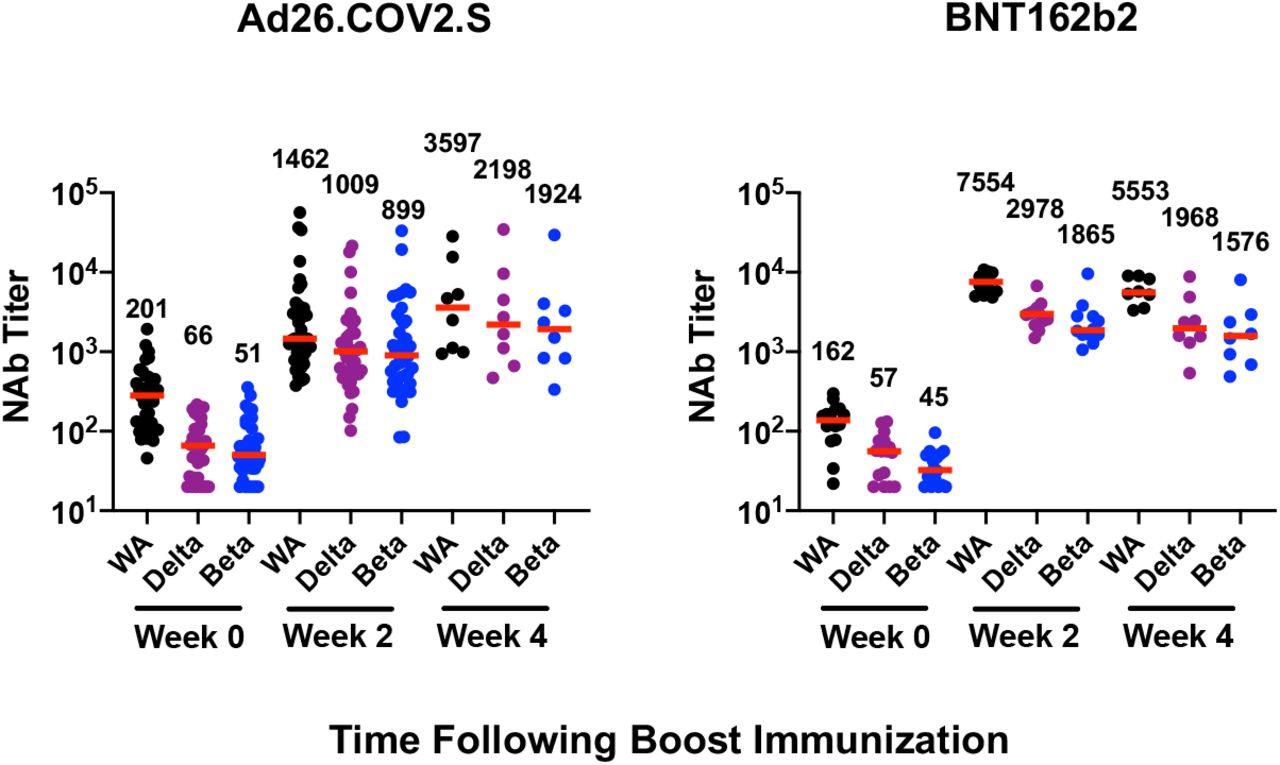
Pseudovirus neutralizing antibody responses were detectable but low at six months following BNT162b2 vaccination, consistent with previous reports showing that serum antibody titers decline sharply following BNT162b2 vaccination1,5. Ad26.COV2.S boosting increased median neutralizing antibody titers to the WA1/2020, delta, and beta variants to 1,462, 1,009, and 899 at week 2 following the boost, respectively, and these titers further increased 2.46-, 2.18-, and 2.14-fold to 3,597, 2,198, and 1,924 at week 4 following the boost (Fig. 1A, Table S2). BNT162b2 boosting increased median neutralizing antibody titers to the WA1/2020, delta, and beta variants to 7,554, 2,978, and 1,865 at week 2 following the boost, respectively, and these titers declined to 5,553, 1,968, and 1,576 at week 4 following the boost (Fig. 1A, Table S2).




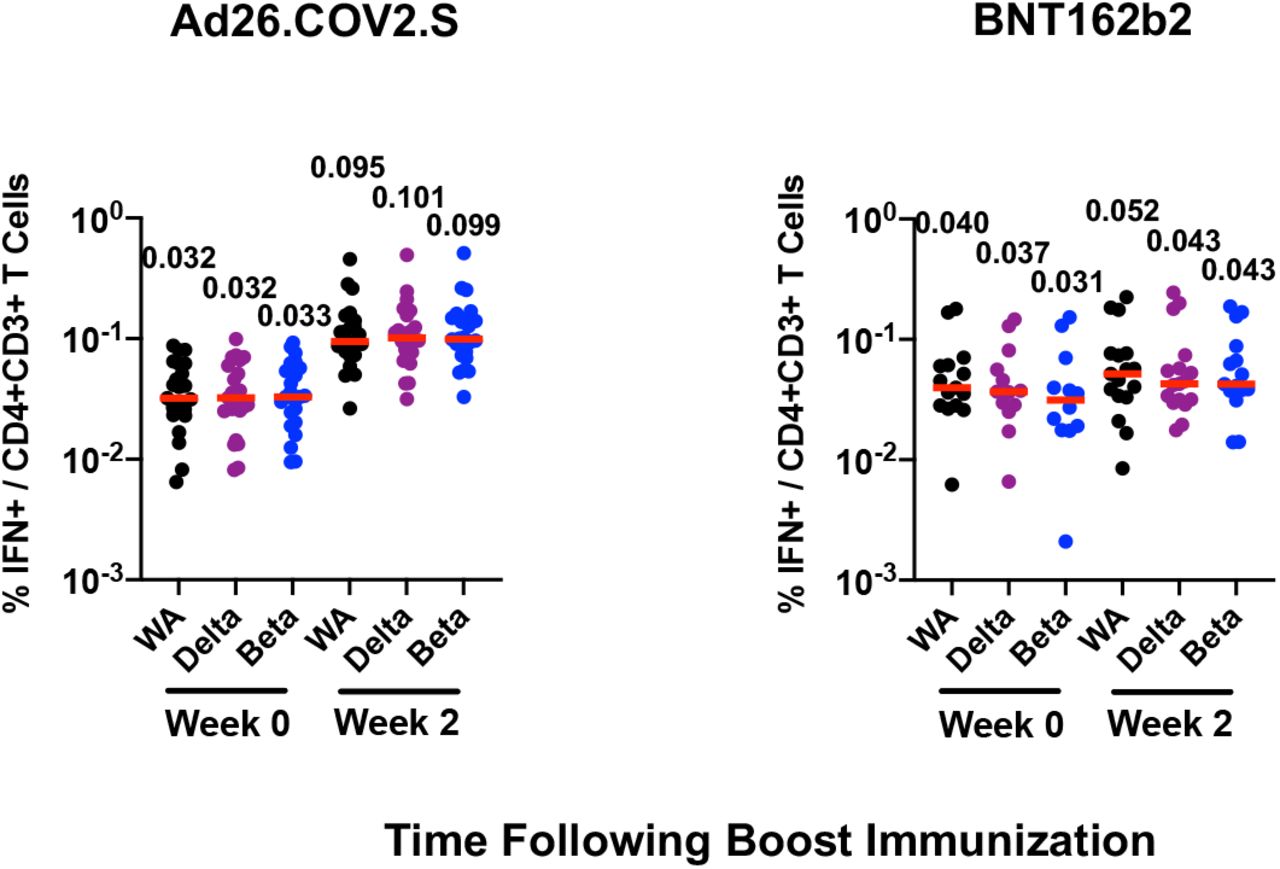
Immune responses at weeks 0, 2, and 4 following boosting BNT162b2 vaccinated individuals with Ad26.COV2.S or BNT162b2. A. Neutralizing antibody (NAb) titers by a luciferase-based pseudovirus neutralization assay. B. Receptor binding domain (RBD)-specific binding antibody titers by ELISA. IFN-γ CD8+ T cell responses (C) and CD4+ T cell responses (D) by intracellular cytokine staining assays. Responses were measured against the SARS-CoV-2 WA1/2020, B.1.617.2 (delta), and B.1.351 (beta) strains. Medians (red bars) are depicted and numerically shown. A subset of samples were available at week 4.
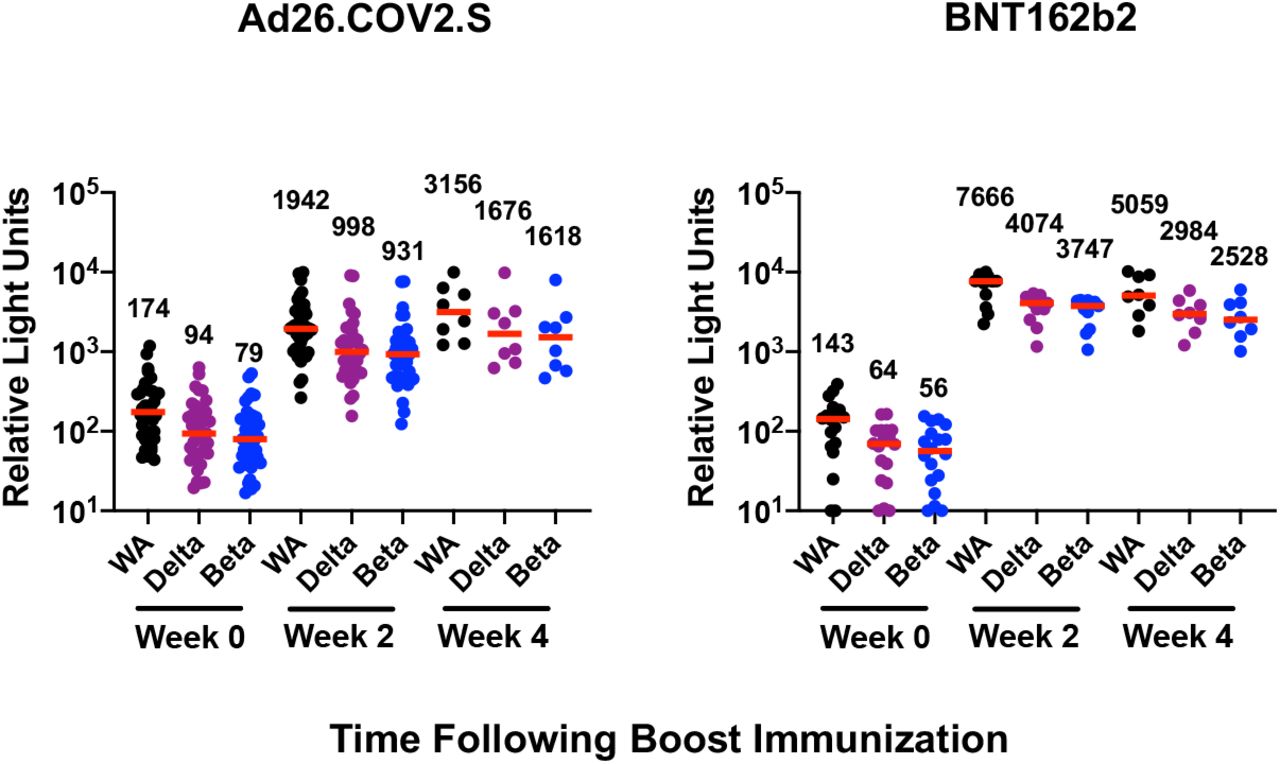
Ad26.COV2.S boosting increased median receptor binding domain (RBD)-specific antibody titers by ELISA to the WA1/2020, delta, and beta variants to 11,264, 10,817, and 5,375 at week 2 following the boost, respectively, and these titers further increased to 16,311, 13,118, and 10,279 at week 4 following the boost (Fig. 1B). BNT162b2 boosting increased median RBD-specific ELISA titers to the WA1/2020, delta, and beta variants to 30,730, 26,398, and 14,725 at week 2 following the boost, respectively, and these titers declined to 14,747, 13,037, and 7,230 at week 4 following the boost (Fig. 1B). Similar trends were observed with an electrochemiluminescence binding antibody assay (ECLA) (Fig. S1). These data show that BNT162b2 boosting led to peak antibody titers at week 2, whereas Ad26.COV2.S led to peak antibody titers at week 4 or later. Binding and neutralizing antibody titers were comparable at week 4 following Ad26.COV2.S and BNT162b2 boosts.
Median S-specific IFN-γ CD8+ T cell responses to the WA1/2020, delta, and beta variants were boosted by Ad26.COV2.S to 0.078%, 0.072%, and 0.083%, respectively, and by BNT162b2 to 0.026%, 0.031%, and 0.018%, respectively (Fig. 1C). Median S-specific IFN-γ CD4+ T cells to the WA1/2020, delta, and beta variants were boosted by Ad26.COV2.S to 0.095%, 0.101%, and 0.099%, respectively, and by BNT162b2 to 0.052%, 0.043%, and 0.043%, respectively (Fig. 1D).
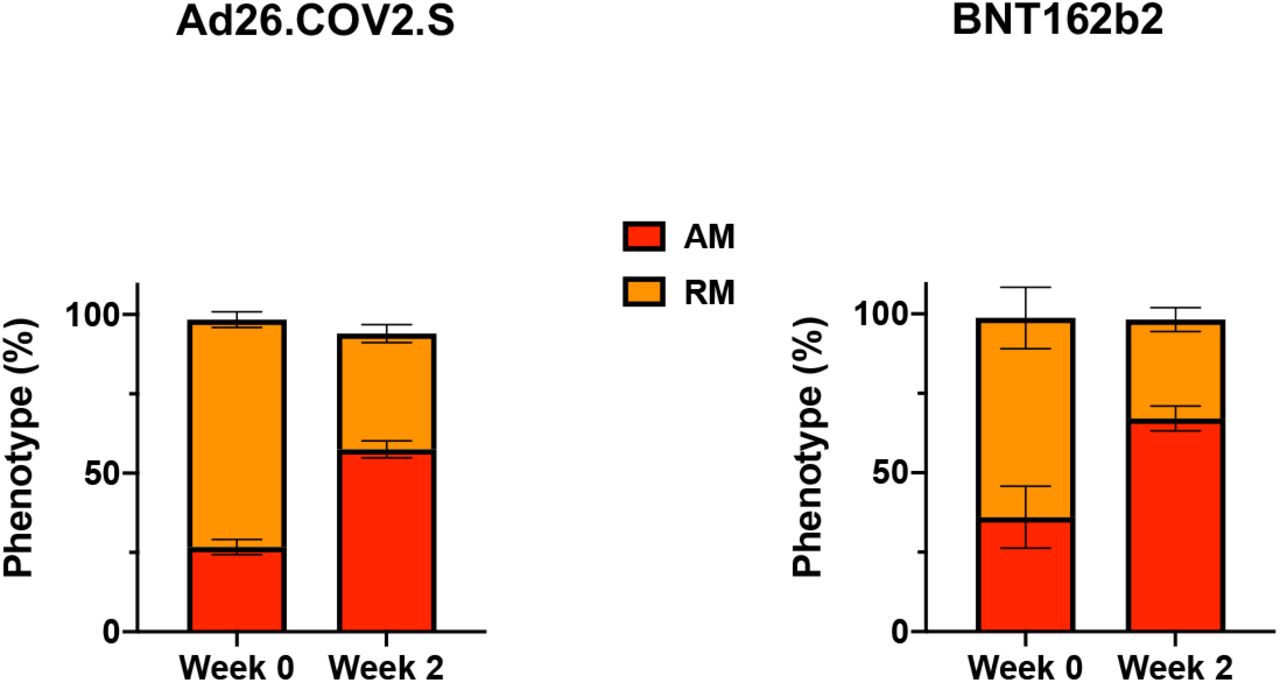
Median RBD-specific memory B cell responses were boosted by Ad26.COV2.S to 0.391% and by BNT162b2 to 0.388% (Fig. S2). RBD-specific B cells exhibited primarily an activated memory phenotype at week 2 following both Ad26.COV2.S and BNT162b2 boosting (Fig. S3).
These data demonstrate that both heterologous Ad26.COV2.S and homologous BNT162b2 increased antibody responses in individuals who were vaccinated at least 6 months previously with BNT162b2. Ad26.COV2.S and BNT162b2 led to similar antibody titers by week 4 following the boost immunization but exhibited different immune kinetics5. Ad26.COV2.S led to greater increases in CD8+ T cell responses than BNT162b2. However, the durability of these immune responses remain to be determined. These data suggest different immune phenotypes following heterologous (“mix-and-match”) compared with homologous boost strategies for COVID-19.
Data Availability
All data produced in the present work are contained in the manuscript
Data sharing
C.S.T, A.Y.C., and D.H.B. had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. All data are available in the manuscript or the supplementary material. Correspondence and requests for materials should be addressed to D.H.B. (dbarouch{at}bidmc.harvard.edu).
Funding
The authors acknowledge Janssen Vaccines & Prevention, NIH grant CA260476, the Massachusetts Consortium for Pathogen Readiness, the Ragon Institute, and the Musk Foundation (D.H.B.). The authors also acknowledge the Reproductive Scientist Development Program from the Eunice Kennedy Shriver National Institute of Child Health & Human Development and Burroughs Wellcome Fund HD000849 (A.Y.C.).
Role of Sponsor
The sponsor did not have any role in design or conduct of the study; collection, management, analysis, or interpretation of the data; preparation, review, or approval of the manuscript; or decision to submit the manuscript for publication.
Conflicts of Interest
DHB is a co-inventor on provisional vaccine patents (63/121,482; 63/133,969; 63/135,182). The authors report no other conflict of interest.
Author Contributions
CST, AYC, DHB: conceptualization, formal analysis, resources, investigation, data curation, writing-original draft, writing-review & editing, visualization, supervision
JL, JY, HW, KAM, XH, CJD, AC, DS: investigation, methodology, data curation, writing-review & editing, supervision
KES, SJV, KJ, JLC, MR, RH, LBR: investigation, resources, methodology, writing-review & editing
TA, JB, BC, SG, MG, ML, OP, HV, CW: resources, methodology, writing-review & editing
Supplementary Methods
Study population
A specimen biorepository at Beth Israel Deaconess Medical Center (BIDMC) obtained samples from individuals who received the BNT162b2 vaccine. The BIDMC institutional review board approved this study (2020P000361). Participants either continued follow-up in the biorepository and were boosted with 30 ug BNT162b2 clinically or were enrolled in the COV2008 study (NCT04999111) and were boosted with 5, 2.5, or 1×1010 vp Ad26.COV2.S. An additional 16 participants who were not enrolled in the BIDMC biorepository were also enrolled in the COV2008 study. The Advarra institutional review board approved the COV2008 study (SSU00161543). All participants provided informed consent. Individuals were excluded if they had a history of SARS-CoV-2 infection, received other COVID-19 vaccines, or received immunosuppressive medications.
Pseudovirus neutralizing antibody assay
The SARS-CoV-2 pseudoviruses expressing a luciferase reporter gene were used to measure pseudovirus neutralizing antibodies as described previously.6,7 In brief, the packaging construct psPAX2 (AIDS Resource and Reagent Program), luciferase reporter plasmid pLenti-CMV Puro-Luc (Addgene) and spike protein expressing pcDNA3.1-SARS-CoV-2 SΔCT were co-transfected into HEK293T cells (ATCC CRL_3216) with lipofectamine 2000 (ThermoFisher Scientific). Pseudoviruses of SARS-CoV-2 variants were generated by using WA1/2020 (Wuhan/WIV04/2019, GISAID accession ID: EPI_ISL_402124), B.1.617.2 (delta, GISAID accession ID: EPI_ISL_2020950), and B.1.351 (beta, GISAID accession ID: EPI_ISL_712096). The supernatants containing the pseudotype viruses were collected 48h after transfection; pseudotype viruses were purified by filtration with 0.45-μm filter. To determine the neutralization activity of human serum, HEK293T-hACE2 cells were seeded in 96-well tissue culture plates at a density of 1.75 × 104 cells per well overnight. Three-fold serial dilutions of heat-inactivated serum samples were prepared and mixed with 50 μl of pseudovirus. The mixture was incubated at 37 °C for 1 h before adding to HEK293T-hACE2 cells. After 48 h, cells were lysed in Steady-Glo Luciferase Assay (Promega) according to the manufacturer’s instructions. SARS-CoV-2 neutralization titers were defined as the sample dilution at which a 50% reduction (NT50) in relative light units was observed relative to the average of the virus control wells.
Enzyme-linked immunosorbent assay (ELISA)
SARS-CoV-2 spike receptor-binding domain (RBD)-specific binding antibodies in serum were assessed by ELISA as described previously.6,7 96-well plates were coated with 2 μg/mL of SARS-CoV-2 WA1/2020, B.1.617.2 (delta), and B.1.351 (beta) RBD protein in 1× Dulbecco phosphate-buffered saline (DPBS) and incubated at 4 °C overnight. After incubation, plates were washed once with wash buffer (0.05% Tween 20 in 1× DPBS) and blocked with 350 μL of casein block solution per well for 2 to 3 hours at room temperature. Following incubation, block solution was discarded and plates were blotted dry. Serial dilutions of heat-inactivated serum diluted in Casein block were added to wells, and plates were incubated for 1 hour at room temperature, prior to 3 more washes and a 1-hour incubation with a 1:4000 dilution of anti– human IgG horseradish peroxidase (HRP) (Invitrogen, ThermoFisher Scientific) at room temperature in the dark. Plates were washed 3 times, and 100 μL of SeraCare KPL TMB SureBlue Start solution was added to each well; plate development was halted by adding 100 μL of SeraCare KPL TMB Stop solution per well. The absorbance at 450 nm, with a reference at 650 nm, was recorded with a VersaMax microplate reader (Molecular Devices). For each sample, the ELISA end point titer was calculated using a 4-parameter logistic curve fit to calculate the reciprocal serum dilution that yields a corrected absorbance value (450 nm-650 nm) of 0.2. Interpolated end point titers were reported.
Electrochemiluminescence assay (ECLA)
ECLA plates (MesoScale Discovery SARS-CoV-2 IgG Cat No: K15463U-2; Panel 13) were designed and produced with up to 10 antigen spots in each well, and assays were performed, essentially as described previously.6,8 The antigens included WA1/2020, B.1.617.2 (delta), and B.1.351 (beta). The plates were blocked with 50 uL of Blocker A (1% BSA in distilled water) solution for at least 30 minutes at room temperature shaking at 700 rpm with a digital microplate shaker. During blocking the serum was diluted to 1:5,000 in Diluent 100. The calibrator curve was prepared by diluting the calibrator mixture from MSD 1:9 in Diluent 100 and then preparing a 7-step 4-fold dilution series plus a blank containing only Diluent 100. The plates were then washed 3 times with 150 μL of Wash Buffer (0.5% Tween in 1x PBS), blotted dry, and 50 μL of the diluted samples and calibration curve were added in duplicate to the plates and set to shake at 700 rpm at room temperature for at least 2 h. The plates were again washed 3 times and 50 μL of SULFO-Tagged anti-Human IgG detection antibody diluted to 1x in Diluent 100 was added to each well and incubated shaking at 700 rpm at room temperature for at least 1 h. Plates were then washed 3 times and 150 μL of MSD GOLD Read Buffer B was added to each well and the plates were read immediately after on a MESO QuickPlex SQ 120 machine. MSD titers for each sample was reported as Relative Light Units (RLU) which were calculated using the calibrator.
Intracellular cytokine staining (ICS) assay
CD4+ and CD8+ T cell responses were quantitated by peptide-stimulated intracellular cytokine staining (ICS) assays, as previously described.6 106 peripheral blood mononuclear cells well were re-suspended in 100 µL of R10 media supplemented with CD49d monoclonal antibody (1 µg/mL) and CD28 monoclonal antibody (1 µg/mL). Each sample was assessed with mock (100 µL of R10 plus 0.5% DMSO; background control), peptides (2 µg/mL), and/or 10 pg/mL phorbol myristate acetate (PMA) and 1 µg/mL ionomycin (Sigma-Aldrich) (100µL; positive control) and incubated at 37°C for 1 h. After incubation, 0.25 µL of GolgiStop and 0.25 µL of GolgiPlug in 50 µL of R10 was added to each well and incubated at 37°C for 8 h and then held at 4°C overnight. The next day, the cells were washed twice with DPBS, stained with aqua live/dead dye for 10 mins and then stained with predetermined titers of monoclonal antibodies against CD279 (clone EH12.1, BB700), CD4 (clone L200, BV711), CD27 (clone M-T271, BUV563), CD8 (clone SK1, BUV805), CD45RA (clone 5H9, APC H7) for 30 min. Cells were then washed twice with 2% FBS/DPBS buffer and incubated for 15 min with 200 µL of BD CytoFix/CytoPerm Fixation/Permeabilization solution. Cells were washed twice with 1X Perm Wash buffer (BD Perm/WashTM Buffer 10X in the CytoFix/CytoPerm Fixation/ Permeabilization kit diluted with MilliQ water and passed through 0.22µm filter) and stained with intracellularly with monoclonal antibodies against IFN-γ (clone B27; BUV395), and CD3 (clone SP34.2, Alexa 700), for 30 min. Cells were washed twice with 1X Perm Wash buffer and fixed with 250µL of freshly prepared 1.5% formaldehyde. Fixed cells were transferred to 96-well round bottom plate and analyzed by BD FACSymphony™ system. Data were analyzed using FlowJo v9.9.
RBD-specific B cell staining
PBMCs were stained with Aqua live/dead dye for 20 min, washed with 2% FBS/DPBS buffer, and cells were suspended in 2% FBS/DPBS buffer with Fc Block (BD) for 10 min, followed by staining with monoclonal antibodies against CD45 (clone HI30, BV786), CD3 (clone UCHT1, APC-R700), CD16 (clone 3G8, BUV496), CD14 (clone M5E2, BV605), CD19 (clone SJ25C, BUV615), CD20 (clone 2H7, PE-Cy5), IgM (clone G20-127, BUV395), IgD (clone IA6-2, PE), CD95 (clone DX2, BV711), CD27 (clone M-T271, BUV563), CD21 (clone B-ly4, PE-CF594), CD38 (clone HB7, BUV805), CD71 (clone M-A712, BV750) and staining with SARS-CoV-2 antigens including biotinylated SARS-CoV-2 (WA1/2020) RBD proteins (Sino Biological), SARS-CoV-2 (WA1/2020) RBD proteins (Sino Biological) labeled with FITC, at 4 °C for 30 min. After staining, cells were washed twice with 2% FBS/DPBS buffer, followed by incubation with BV650 streptavidin (BD Pharmingen) for 10 min, then washed twice with 2% FBS/DPBS buffer. After staining, cells were washed and fixed by 2% paraformaldehyde. All data were acquired on a BD FACSymphony flow cytometer. Subsequent analyses were performed using FlowJo software (BD Bioscience, v.9.9.6). For analyses, in singlet gate, dead cells were excluded by Aqua dye and CD45 was used as a positive inclusion gate for all leukocytes. Within class-switched B cell population gated as CD19+ IgM-IgD-CD3-CD14-CD16-, SARS-CoV-2 RBD-specific B cells were identified as double positive for SARS-CoV-2 (WA1/2020) RBD proteins labeled with different fluorescent probes. The SARS-CoV-2-specific B cells were further distinguished according to CD21/CD27 phenotype distribution as previously described 9.
Statistical analysis
Descriptive statistics were calculated using GraphPad Prism 8.4.3, (GraphPad Software, San Diego, California). Data are presented as median with interquartile range (IQR).
Characteristics of the study population
Ratio of NAb titers (week 4 / week 2)
Supplementary Figure Legends

Binding antibody titers at weeks 0, 2, and 4 following boosting BNT162b2 vaccinated individuals with Ad26.COV2.S or BNT162b2 were assessed by electrochemiluminescence binding antibody assay (ECLA). Responses were measured against the SARS-CoV-2 WA1/2020, B.1.617.2 (delta), and B.1.351 (beta) strains. Medians (red bars) are depicted and numerically shown. A subset of samples were available at week 4.
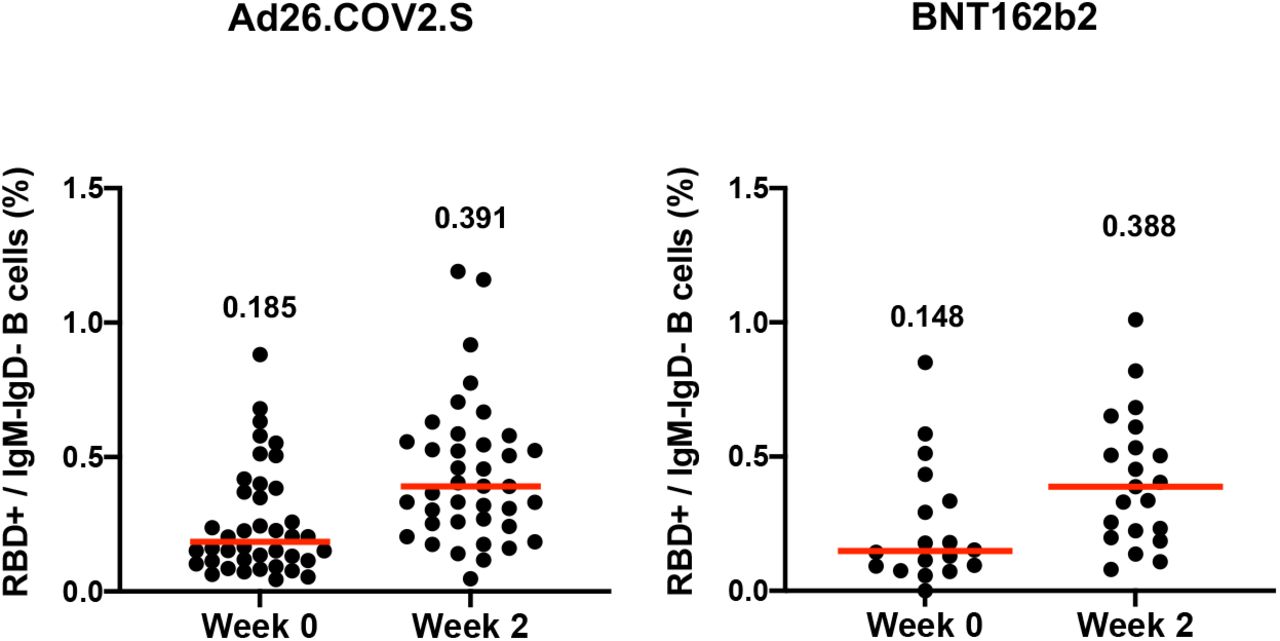
Receptor binding domain (RBD)-specific memory B cells at weeks 0 and 2 following boosting BNT162b2 vaccinated individuals with Ad26.COV2.S or BNT162b2. Medians (red bars) are depicted and numerically shown.

Phenotype of RBD-specific memory B cells at weeks 0 and 2 following boosting BNT162b2 vaccinated individuals with Ad26.COV2.S or BNT162b2. Red, activated memory (AM) B cells. Orange, resting memory (RM) B cells.
Acknowledgements
The authors thank the study participants, the Center for Virology and Vaccine Research, and the Department of Obstetrics and Gynecology for enrollment, collection, and processing samples for the BIDMC COVID-19 Biorepository. We thank MesoScale Discovery for providing the kits for the ECLA multiplexing kits used in this study.
Footnotes
↵* Co-first author
Authors for Print Edition C. Sabrina Tan, M.D., Ai-ris Y. Collier, M.D. and Dan H. Barouch, M.D., Ph.D.; Center for Virology and Vaccine Research, Beth Israel Deaconess Medical Center, Boston, MA, USA




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}