
Abstract
Prisons are high-incidence settings for tuberculosis around the world, yet the contribution of spillover from prisons in driving community epidemics has not been quantified. We whole genome sequenced 1,152 M. tuberculosis isolates from participants diagnosed with tuberculosis within prisons and in the community in Mato Grosso do Sul, Brazil from 2014 to 2019. By integrating timed phylogenies and detailed location data, we reconstructed probabilistic transmission histories. M. tuberculosis sequences from incarcerated and non-incarcerated people were closely phylogenetically related. We found that 57% of recent community-wide tuberculosis cases were attributable to transmission from individuals with an incarceration history, 2.6% of the population. Further, we find genomic evidence that the prison system disseminates M. tuberculosis genotypes through frequent transfers across the state. This population-wide genomic transmission reconstruction framework can be applied to identify key environments amplifying infectious disease transmission to prioritize public health interventions.
Teaser Prison spillover plays an outsized role in community tuberculosis transmission.
Introduction
Tuberculosis remains one of the leading causes of death by an infectious disease and led to 1.4 million deaths in 2019.(1) Locating where M. tuberculosis transmission occurs is critical for public health interventions, yet in high-incidence settings, where most tuberculosis is transmitted remains unknown.(1–6) Prisons, hospitals, or mines are known to be high-risk environments(7–9) and may have epidemiological impacts that transcend their walls, serving as institutional amplifiers of infection.(10, 11) Identifying institutional amplifiers of tuberculosis is of particular concern in Central and South America, where the incarcerated population has increased by over 200% since 2000 and 11% percent of tuberculosis cases now occur among incarcerated people, who comprise <1% of the total population.(12)
The full impact of increasing incarceration rates on the tuberculosis epidemic in the Americas and elsewhere has been difficult to quantify. Many people are infected in prison, but only diagnosed later.(11, 13, 14) Further, spillover may occur when incarcerated people, prison staff, or visitors are infected within high-incidence prison environments and transmit M. tuberculosis onwards in communities outside prison. Previous studies have found that the excess risk of tuberculosis within prisons extends to surrounding communities(15, 16) and that M. tuberculosis genotypes responsible for jail and prison outbreaks are also found in the surrounding communities.(13, 15, 17, 18)
Pathogen genomes can be powerfully harnessed to infer high-resolution transmission histories, including who-infected-whom.(19) However, studies have not yet combined M. tuberculosis genomic and epidemiologic data to reconstruct transmission linkages between potential institutional amplifiers and the community, nor have they estimated the fraction of community transmission attributable to such spillover. To quantify the role of prisons in population-wide tuberculosis transmission, we conducted prospective genomic surveillance for tuberculosis in the Brazilian state of Mato Grosso do Sul and used full M. tuberculosis genomes, combined with detailed meta-data on incarceration history, to reconstruct tuberculosis transmission chains spanning prisons and the community.
Results
Tuberculosis and incarceration trends in Mato Grosso do Sul1
Brazil’s incarcerated population grew by 107% (from 361,402 to 748,009) from 2005 to 2019, an increase closely paralleled in the Central West state of Mato Grosso do Sul, where the incarcerated population has grown by 115% (7,891 to 16,976) over the same period (Fig. 1a-c). In 2019, prisons in the state were at 203% occupancy. While the state’s notification rate of new and retreatment tuberculosis in the general population (38 per 100,000) is similar to Brazil’s national tuberculosis notification rate (40 per 100,000) in 2019,(20) the notification rate within prisons was 43 times as high (1,666 per 100,000), again similar to Brazil’s national notification rate within prisons (1,596 per 100,000) in 2019 (Fig. 1d,e).

(a) Map of Brazil with states colored by the 2019 incarceration rate with Mato Grosso do Sul outlined in black. (b) Map of Mato Grosso do Sul state with the cities with passive surveillance only and where active surveillance was additionally conducted within prisons. (c) The incarcerated population size in Mato Grosso do Sul grew by 142% from 2005 to 2020, increasing from 7,891 to 19,065 people. (d) Mato Grosso do Sul state’s annual new and retreatment tuberculosis notifications, colored by incarceration status and (e) the notification rate per 100,000 people for the incarcerated population and non-incarcerated populations from 2009 to 2019. The y-axis is log-scaled. The mean tuberculosis notification rate was more than 46 times greater among the incarcerated population compared to the non-incarcerated population. The green and yellow bars (d) and (e) indicate the period of active surveillance in prisons in Campo Grande and Dourados respectively.
Population-based genomic surveillance across Mato Grosso do Sul state
From 2014 to 2019, 3,491 new and retreatment cases of tuberculosis were reported in the Campo Grande and Dourados, the two largest cities in Mato Grosso do Sul, Brazil. Of these, 1,249 had positive cultures and we sequenced 787 M. tuberculosis isolates. We sequenced additional isolates from three other cities in the state and from earlier years in Dourados for a total of 1,090 isolates from unique tuberculosis notifications (Fig. 1b). Whole genome sequences (WGS) for 1,043 isolates met our coverage and quality criteria (Methods). We excluded 10% (108/1043) of isolates with mixed infection, resulting in 935 high-quality genomes from 935 unique tuberculosis episodes from 918 individuals.
Of the 935 isolates in our analyses, 50% (465/935) were incarcerated; 16% (150/935) were from patients who were formerly incarcerated; and 34% (320/935) of the study population did not have an incarceration history at the time of tuberculosis notification. Among those who did not have an incarceration history, 32 people reported contact with incarcerated individuals. Additional population characteristics are in Table S1. Isolates were largely from M. tuberculosis lineage 4 and predominantly fell into three sublineages: lineage 4.1, 4.3, and 4.4 (Fig. 2). Overall, we found a low prevalence of antibiotic resistance across isolates, with 93.7% of isolates susceptible to all antibiotics (Fig. 2).
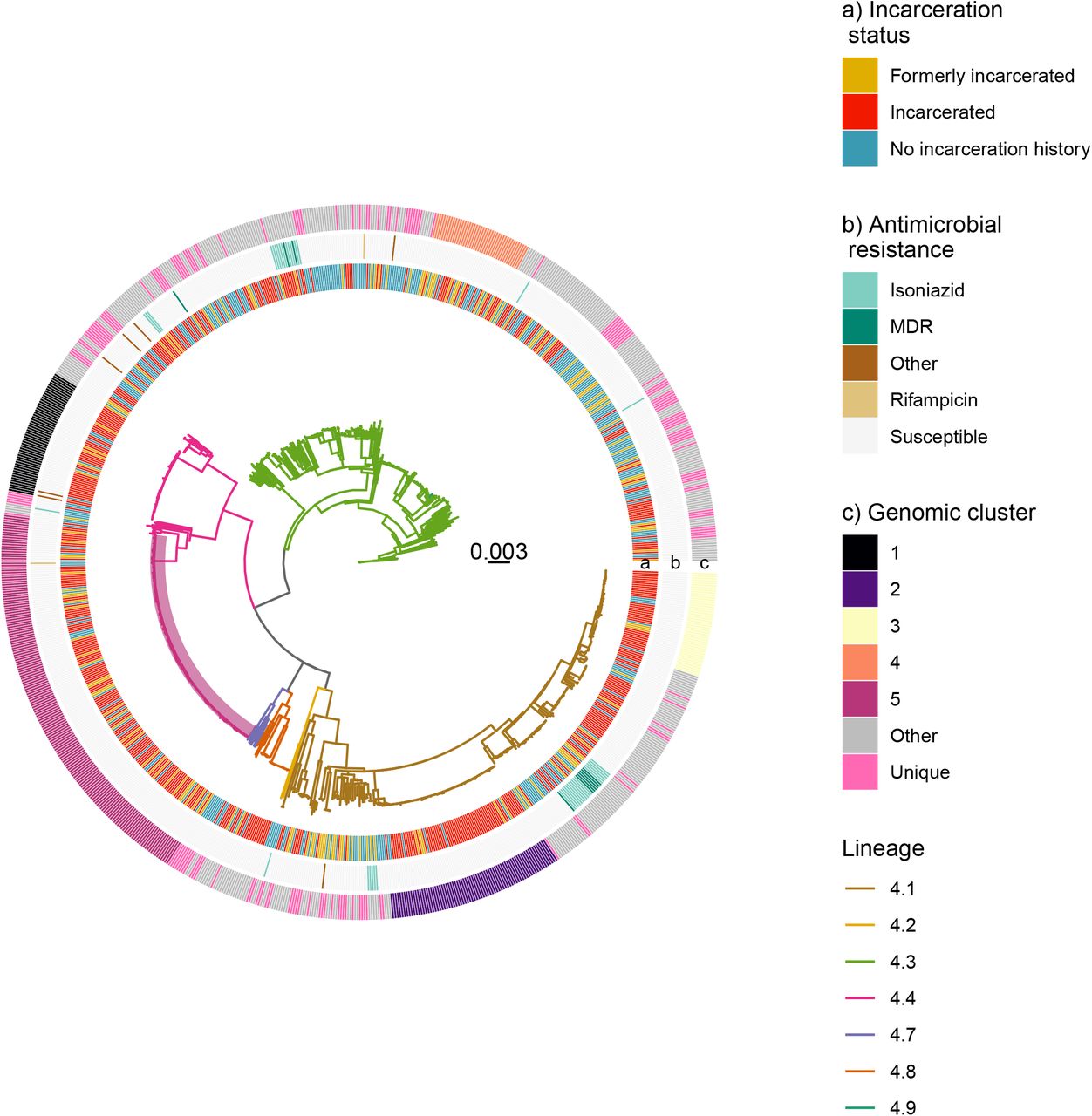
A maximum likelihood phylogeny of 932 tuberculosis isolates from Lineage 4 inferred from a multiple sequence alignment of 19,753 SNPs. Branch lengths are in units of substitutions per site. From the inside, rings are colored by incarceration status, antimicrobial resistance prediction, and genomic cluster (Methods). “Other” resistance indicates resistance to at least one antibiotic other than isoniazid or rifampicin. Branches are colored by sublineage. The clade containing the largest predicted genomic cluster (Cluster 5), including 170 isolates, is highlighted in purple.
Phylogenetic structure of M. tuberculosis from prisons and the community
A maximum likelihood phylogeny (Fig. 2) demonstrates that M. tuberculosis isolates sampled from incarcerated and non-incarcerated people do not form distinct clades and are closely phylogenetically related. Terminal branch lengths, a proxy for the degree of recent transmission,(21) are significantly longer for isolates from people with no incarceration history at the time of tuberculosis notification (9.4 × 10−4 substitutions/site) than isolates from incarcerated individuals (2.2 × 10−4; two-sample t-test p-value = 1.14 × 10−7) or formerly incarcerated individuals (3.8 × 10−4; p = 1.6 × 10−4), suggesting that individuals with an incarceration history were more likely to be recently infected (Fig. S1).
Genetic clustering of M. tuberculosis from prisons and the community
To identify potential clusters of recent transmission, we applied a commonly used 12-SNP threshold,(22) including all isolates within the threshold distance of at least one other clustered isolate. Eighty-three percent (777/935) of the isolates fell into 84 genomic clusters (each including 2 to 170 isolates), providing evidence that notifications were largely attributable to recent, local transmission rather than travel-associated importation or re-activation of genetically distinct latent infections (Fig. 2). Isolates from incarcerated people were more frequently clustered (93.3%, 434/465), than those from formerly incarcerated (86.0%, 129/150; p <0.0001), or never incarcerated people (66.9%, 214/320; p <0.0001), again suggesting more recent transmission within prisons.
We predicted that if prison and community-associated epidemics were distinct, isolates from the community would be most closely related to and cluster with other isolates from the community and vice versa. Of the 46 clusters with three or more isolates, including 703 participants, 40 included isolates from community members who had no reported incarceration history. Of those potential transmission clusters, 85.0% (34/40) included isolates from people who were currently or previously incarcerated.
We found a similar pattern of clustering of isolates from community members and individuals with an incarceration history when using an alternative 5-SNP threshold for genomic clustering(23). With this threshold, of the 80 clusters with three or more isolates, 56 included isolates from people with no incarceration history and 78.6% (44/56) also included isolates from people with an incarceration history.
A M. tuberculosis clone spans prisons and the community across the state
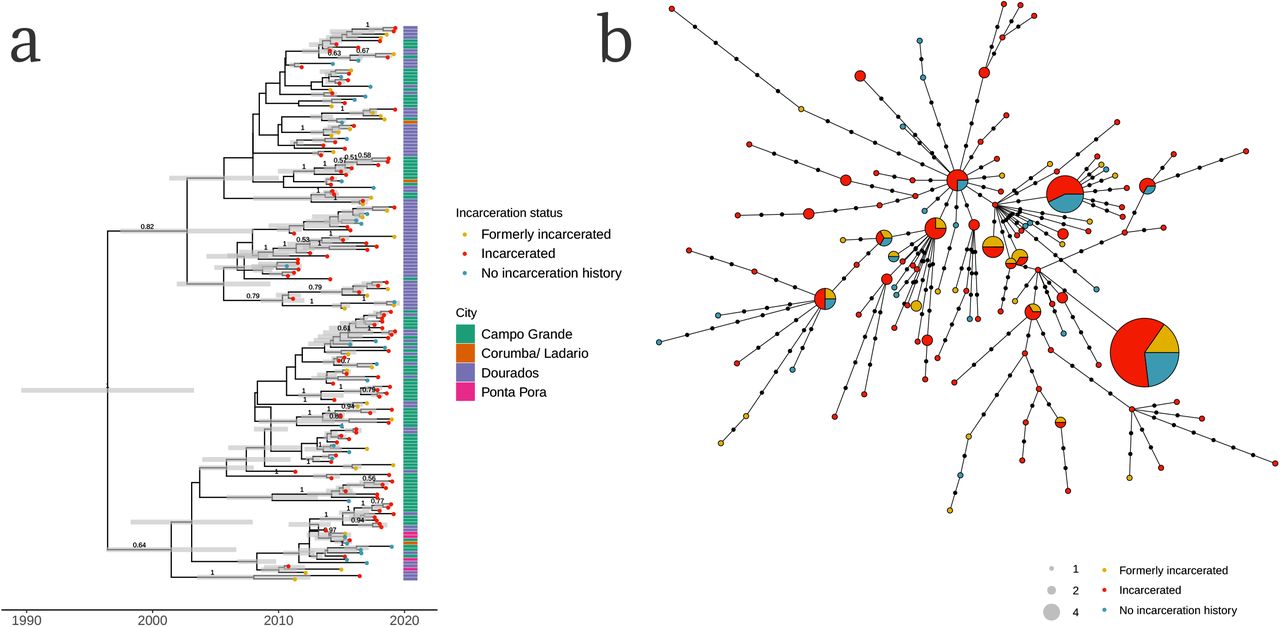
The largest potential transmission cluster, including 170 isolates sampled from September 2010 to April 2019, was distributed across the state, including cases found across state’s two largest cities, Campo Grande and Dourados, as well as the smaller cities Corumbá (on the Bolivian border) and Ponta Porã (on the Paraguayan border) (Fig. 1b, Fig. 3a). The cluster had a most recent common ancestor in 1996 (95% HPD: 1989-2003; Fig. 3a), indicating that the drug-susceptible clone had circulated for approximately 23 years before the most recent samples were collected. 103 isolates were from people incarcerated at the time of diagnosis, 35 from people formerly incarcerated, and 32 from people with no incarceration history at the time of tuberculosis notification.
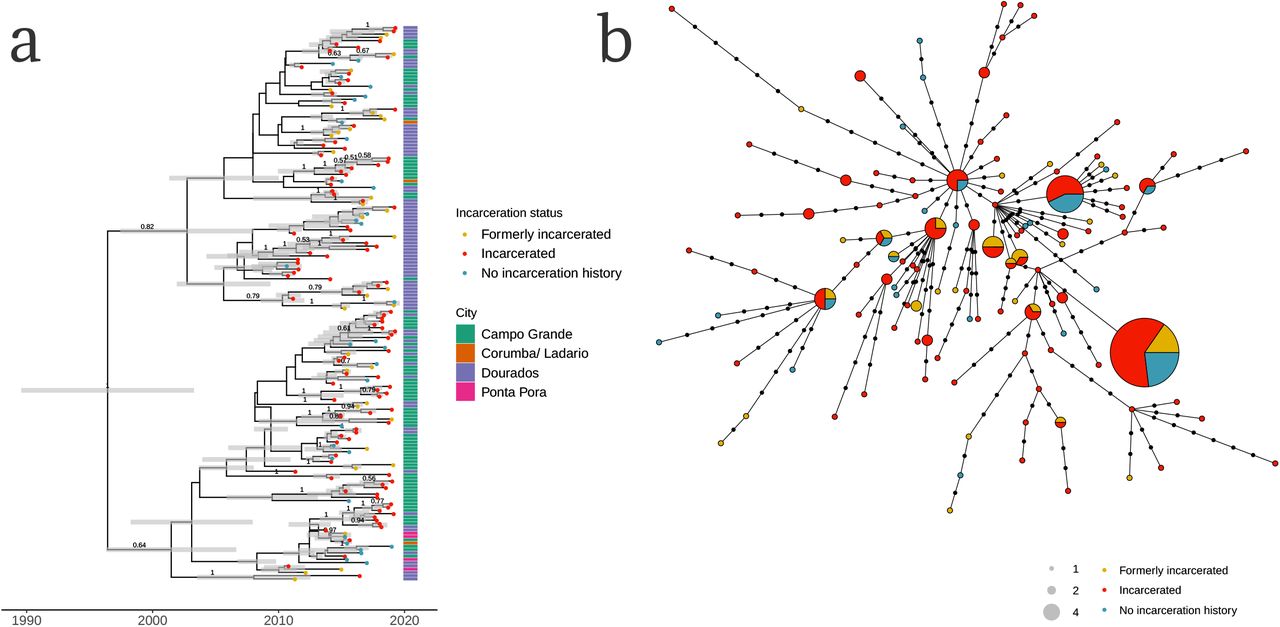
(a) A Bayesian time-calibrated phylogeny of the largest sampled genomic cluster of 170 isolates. The cluster emerged approximately 23 years before the most recently sampled isolate, with a most recent common ancestor in 1996 (95% HPD: 1989-2003). Tip point color indicates patient’s incarceration status at the time of tuberculosis notification. Annotation bar colors indicate city. Grey error bars indicate the 95% Bayesian highest posterior density intervals for node date. Clade posterior support values are shown on the middle of branches for clades with posterior support > 0.5. (b) A haplotype network of the single largest genomic cluster, including 170 isolates. Nodes represent unique haplotypes and are scaled to size. Points along branches indicate SNP distances between haplotypes. Node color indicates incarceration status at the time of diagnosis.
As observed in other clusters, isolates from people who are currently and formerly incarcerated were closely genetically related—and often, identical—to isolates from people who were not incarcerated at the time of notification, suggesting they are linked through recent transmission (Fig. 3b).
We hypothesized that the criminal justice system’s frequent transfer of people between prisons and jails could disseminate TB across the state. Brazil’s national criminal law defines different “regimes” or stages of incarceration(30), which may facilitate the potential for spillover and dissemination of infection. People are incarcerated under closed regimes, within prisons; semi-open regimes, in which people may work outside prisons and return at night; and open regimes, under which people serve sentences outside of prison, but are required to make periodic court appearances.(30) We analyzed movements in the state’s incarceration database and found that the average duration of incarceration was 230 days in a prison and 25 days in a jail. The incarceration database documents 7,982 mean yearly transfers between prisons (including closed and semi-open prisons) from 2015-2018 in a prison population of 17,221 in August 2018, including frequent transfers between cities across the state (Fig. 4a).

(a) Incarceration locates people within highly connected contact networks. Map of Mato Grosso do Sul state with orange points indicating the 21 prisons which most frequently transfer people between prisons. Edges are colored by the mean number of yearly transfers made between prisons from 2015-2018, including transfers in both directions. Edges were drawn between prison pairs with more than 10 transfers in the study period. Transfers between prisons within a city are not shown. (b) An M. tuberculosis clone (depicted in Figure 3) spreads throughout multiple prisons, jails, and the community. Each line represents an individual patient infected with an isolate in the largest genomic cluster, ordered by notification date. Line color indicates patient location, determined by matching patient names with the state incarceration database (Methods). Points indicate TB notification dates; point color indicates patient location at the time of TB notification. Individuals with notification dates after December 31, 2018 are not shown.
To investigate the role of the criminal justice system in disseminating infection, we tracked the spread of the largest sampled M. tuberculosis cluster (Fig. 3) across prisons in Mato Grosso do Sul (Fig. 4b). From early 2011 to 2014, sampled isolates from the cluster were concentrated in the maximum security prison in Dourados, after which it was identified within two prisons in Campo Grande and then exported to prisons and jails in other cities. The cluster was not contained within the state’s prison network; notified tuberculosis cases among community members outside of prison occurred over the duration of the clusters’ state-wide spread.
Transmission trees reveal who-infected-whom, where, and when transmission occurred
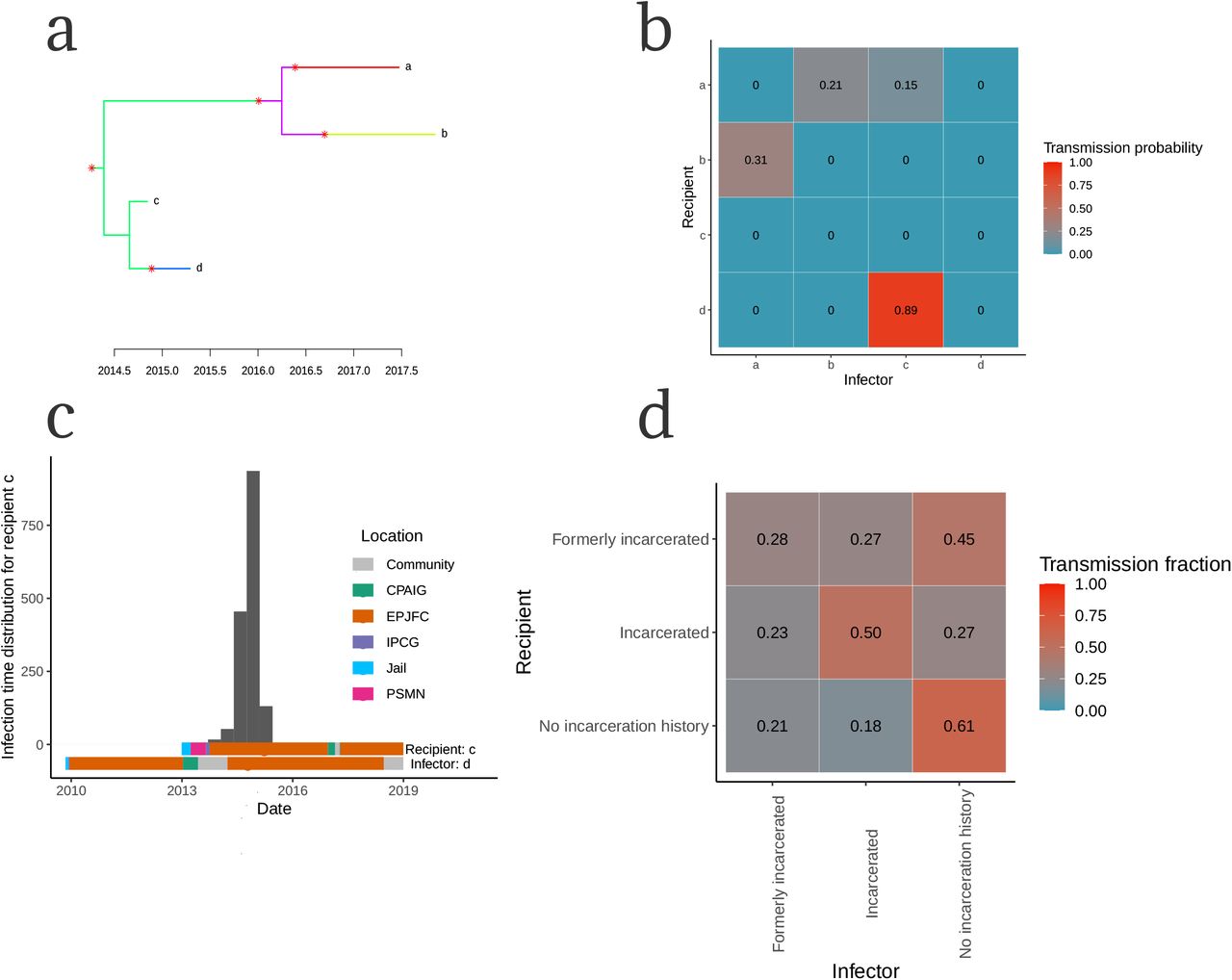
We further investigated genomic clusters by integrating timed phylogenies with epidemiological information to infer transmission trees, allowing us to reconstruct not only transmission linkages of who-infected-whom, but also locate when and where transmission occurred. Reconstructed transmission linkages revealed frequent transmission within prisons and spillover from prisons to the community. For example, we inferred likely transmission (89% posterior probability) between two individuals (Fig. 5a,b) and occurred between July 2014 and July 2015 with 94% posterior probability (Fig. 5c). Both the recipient and predicted infector were incarcerated in the same prison in Campo Grande at the time of the recipient’s tuberculosis notification and that transmission occurred within this prison with a posterior probability of 97%.
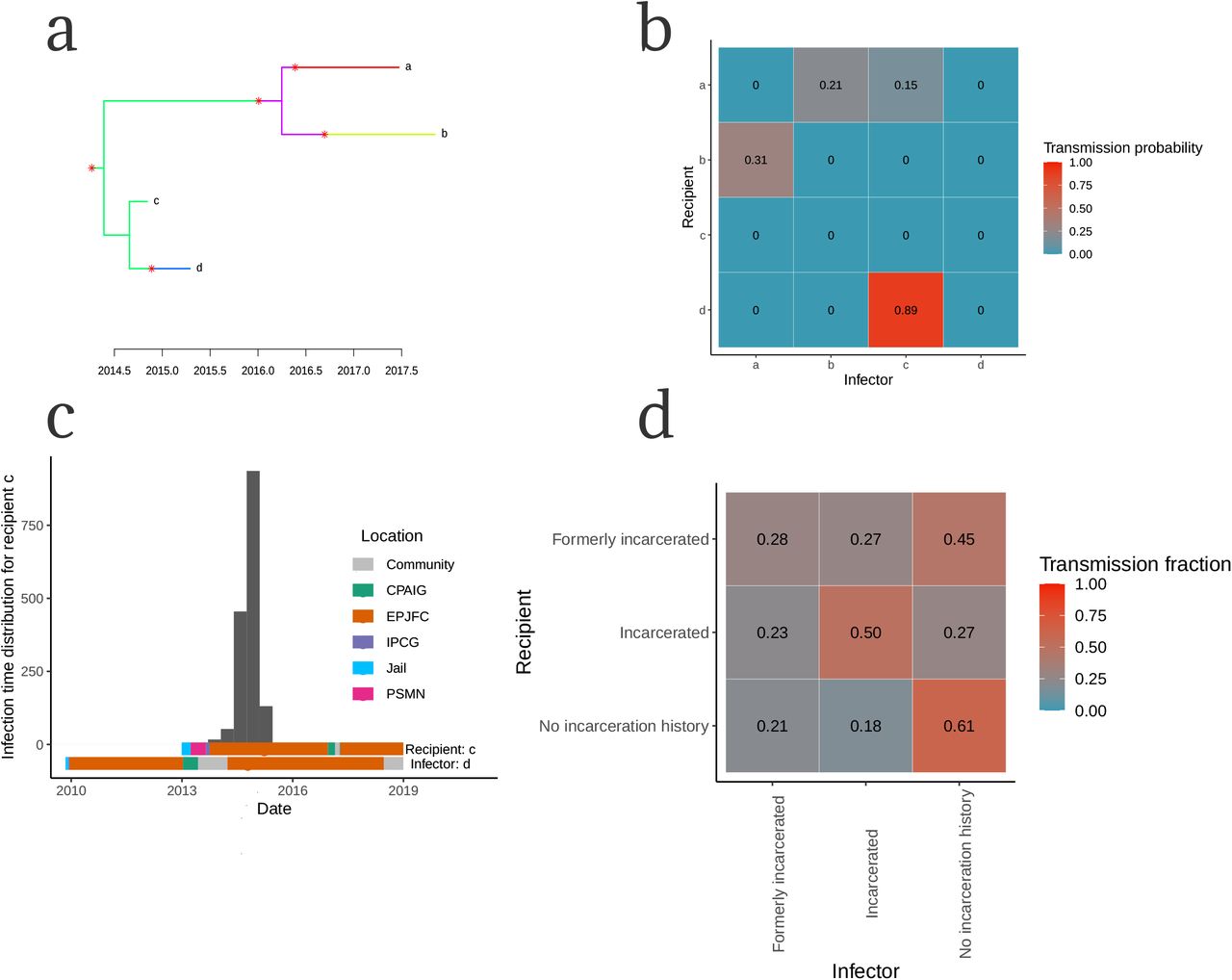
We integrated M. tuberculosis phylogenetic trees with epidemiologic data to infer probable transmission linkages among sampled patients as demonstrated by an example cluster (a-c). (a) Transmission histories can be represented by a phylogenetic trees with branches colored by host in which tips represent sampled isolates and transmission events are indicated by asterisks. (b) We summarized sampled transmission trees into who-acquired-infection-from-whom matrices in which rows represent individual recipients, columns represent individual infectors, and each cell is colored by pairwise transmission probability. (c) The posterior distribution of infection times (grey histogram) for individual d with bars below representing location information for individual d and individual c, the predicted infector. Bars are colored by location over time and circles indicate date of tuberculosis notification. (d) The population-wide WAIFW matrix describing the proportions of observed transmissions to each recipient population from each infector population, corrected for genomic sampling (Methods). Rows represent recipient populations, columns represent infector populations, and cells indicate the proportion of total infections to each recipient population from each infector population defined by their incarceration status at the time of transmission and corrected for sampling rate.
Spillover from prisons plays a disproportionate role in community transmission
To estimate the total contribution of different populations to community-wide tuberculosis transmission, we inferred transmission trees for all clustered isolates. We identified the location of both the infector and recipient over the distribution of likely infection times, allowing us to determine location-specific transmission probabilities. Finally, we summed transmission probabilities over all potential transmission pairs by the incarceration status of both the infector and recipient at the time of transmission, giving a population-wide WAIFW matrix (Fig. 5d). Because we conducted active surveillance inside prisons and not in the general population, we corrected pairwise transmission probabilities to account for higher genomic sampling proportion of incarcerated populations (Fig. S2, Methods).
The population-wide WAIFW matrix reveals that a small proportion of the state’s population, 0.84% (22,706 incarcerated people of a total population of 2,713,147 in 2017), were associated with 18% of observed transmissions to people with no incarceration history (Fig. 5d). When additionally including people who were previously incarcerated, 2.6% of the population (71,703 of 2,713,147) were associated with 39% of the transmission to people with no incarceration history (Fig. 5d), demonstrating the outsized impact of prisons on tuberculosis transmission in the state. In total, we found that 57% of all infections captured in our study (including incarcerated, formerly incarcerated and never incarcerated individuals), including 46% of infections in the community (among formerly incarcerated and never incarcerated individuals), were attributable to transmission from incarcerated or formerly incarcerated individuals, who comprise a small fraction of the state’s population.
Transmission inferences are robust to epidemiological priors
We conducted a sensitivity analysis to determine the effect of epidemiological priors (Table S2) on transmission inferences for the largest genomic cluster (170 isolates). Transmission probabilities inferred for pairs of individuals were closely correlated across generation time priors (Pearson’s r: 0.94 – 0.98, Fig. S3) and sampling time priors (Pearson’s r: 0.84 – 0.98, Fig. S4), although the total number of observed transmission events was strongly influenced by both priors (Figure S5). This suggests that, as expected, epidemiological priors impact the magnitude of inferred transmission probabilities rather than the identity of predicted transmission pairs. Further, alternative priors did not strongly influence the population-wide WAIWF matrix for the largest genomic cluster (Fig. S6). The fraction of transmission to those with no incarceration history attributable to spillover (from infectors who are currently or previously incarcerated) in the largest cluster ranges from 63.5% (medium sampling and generation times) to 72.3% (short generation time and long sampling time).
Discussion
Here, we find genomic evidence that spillover from a key, high-risk environment can drive tuberculosis transmission risk in the broader population. Controlling tuberculosis thus urgently requires reducing risk created by institutional amplifiers, which have outsized effects on the general population.(11) Our population-level transmission reconstruction framework can be applied to identify other high-risk environments or populations and quantify their contribution to community tuberculosis transmission, informing targeted public health interventions.(11)
The disproportionate role that prisons play in driving community tuberculosis transmission is not unique to Brazil. Incidence of tuberculosis in prisons is extremely high globally.(7, 8) The rapid growth in incarceration rates across Central and South America, in particular, puts an increasing population at heightened risk of tuberculosis—risk that we have found can extend into neighboring communities. Although the prevalence of antibiotic resistance is low in Central West, Brazil, prisons have also amplified drug-resistant tuberculosis in Russia(24) and elsewhere. Further, the role of prisons and other detention centers as disease reservoirs is not unique to tuberculosis. Previous studies have identified spillover of meningococcal disease(25) and SARS-CoV-2(26) from prisons to the community.
Our results emphasize that reducing the extraordinarily high transmission risk created by prison environments is an urgent priority. Public health programs need to work to reduce the extreme overcrowding within prisons; improve the unsanitary, often inhumane conditions of incarceration; and expand access to primary healthcare and nutrition. Tuberculosis control programs should expand routine active screenings during(11, 27) and following incarceration so that cases can be diagnosed early and linked to treatment. Individuals leaving prison are at heightened risk of tuberculosis and may be a particularly vulnerable population. Further, preliminary evidence suggests that isoniazid protective therapy is protective against tuberculosis infection and should be considered as a protective intervention in high-risk prison environments.(28)
Reducing this excess burden of disease will require work that extends beyond biomedical interventions. The most direct way to mitigate the excess tuberculosis risk created by prisons is for governments to identify alternatives to detention so that incarceration is used only as a last resort. Prison releases have already begun as a means of reducing infectious disease transmission risk:(29) in an attempt to reduce the risk of COVID-19 in prisons, Brazil released more than thirty thousand people early by mid-2020.(30)
Our study has several limitations. First, while we were able to enroll a majority of M. tuberculosis isolates from incarcerated people with tuberculosis in the city of Dourados, overall sampling of the state’s ongoing tuberculosis epidemic was incomplete. To minimize sampling bias, we determined genomic sampling probabilities and adjusted transmission probabilities to account for over-sampling of prisons. Our probabilistic approach allows us to account for uncertainty in transmission linkages; however, we still cannot infer the identity or incarceration history of unsampled people who contribute to transmission. Terminal branch lengths may similarly be affected by sampling bias or pathogen population size and are thus an imperfect proxy of recency of transmission.
Second, transmission trees reflect information from timed M. tuberculosis phylogenies and epidemiological priors. It is difficult to distinguish genomic sampling proportion from generation time and sampling time distributions. Because we conducted transmission inference in a tuberculosis-endemic setting, rather than in a discrete outbreak, our rate of genomic sampling was lower than in some previous tuberculosis transmission studies. To reduce uncertainty in sampling proportion, we conducted transmission inference on recent subtrees (MRCA > 2012) for which we had sampled a greater proportion of isolates. To investigate the influence of epidemiological priors on transmission inferences, we conducted a sensitivity analysis (Table S2) and found that our conclusions were largely unchanged (Supplementary Text, Figs. S3-S6).
Third, we conducted whole genome sequencing of M. tuberculosis isolates cultured from sputum and generated single consensus genomes from each participant. Excluding within-host diversity could lead to some incorrect transmission inferences. For example, if we sampled an M. tuberculosis genome that was genetically distant from the genome that was transmitted, we could incorrectly exclude transmission.
Finally, we employed an existing bioinformatic approach to identify M. tuberculosis variants and excluded repetitive genomic regions as is common practice.(31, 32) Future studies that leverage within-host M. tuberculosis diversity and/or include the diverse PE/PPE genes could provide greater resolution and reduce the uncertainty of transmission inferences.(33)
In this study, we present genomic evidence that prisons act as tuberculosis reservoirs in Central West, Brazil. The dramatic expansion of incarceration in recent decades has put an increasing population at extremely high risk of tuberculosis; this risk extends to surrounding communities. Reducing the excess tuberculosis transmission risk within prisons and other detention centers is an urgent public health priority. Our findings additionally highlight the potential for pathogen genomic surveillance to identify other key environments or core populations with a disproportionate role in pathogen transmission and guide public health priorities.
Materials and Methods
Study design
We conducted population-based tuberculosis surveillance in the state of Mato Grosso do Sul in Central West Brazil from January 2014 through May 2019. Surveillance included active screening in three of the largest prisons in the state as well as ongoing passive surveillance focused on the two largest cities in the state, Campo Grande and Dourados, and three cities at the state’s border with Paraguay and Bolivia (Fig. 1a,b; Supplementary Methods). All participants provided written consent, and this study was conducted with the approval of the Research Ethics Committee from the Federal University of Grande Dourados, Federal University of Mato Grosso do Sul and National Research Ethics Committee (CONEP) (CAAE 37237814.4.0000.5160, 2676613.3.1001.5160, and 26620619.6.0000.0021) and Stanford University Institutional Review Board (IRB-40285).
Incarceration history
To investigate the incarceration history of tuberculosis patients more closely, we obtained permission from the Mato Grosso do Sul state prison administration agency, Agência Estadual de Administração do Sistema Penitenciário, to access a database of all prison entries, exits, and transfers within the criminal justice record system, Sistema Integrado de Gestão Operacional, from January 1, 2005 through December 31, 2018. Brazil’s national criminal law defines different “regimes” or stages of incarceration(34), which may facilitate the potential for spillover of infection. People are incarcerated under closed regimes, within prisons; semi-open regimes, in which people may work outside prisons and return at night; and open regimes, under which people serve sentences outside of prison, but are required to make periodic court appearances.(34)
Whole genome sequencing
We sequenced whole genomes from cultured isolates on an Illumina NextSeq (2 × 151-bp). Sequence data is available on the Sequence Read Archive (SRA), in BioProject PRJNA671770. We applied variant calling methods closely following those described in Menardo et al.(31) to be consistent with the methods used for molecular clock estimation (Supplementary Methods).
Phylogenetic and Bayesian evolutionary analysis
We fit maximum likelihood trees with RAxML-ng 1.0.1.(35) For each cluster including three or more isolates, we fit Bayesian trees with BEAST 2.6.2(36) with a strict clock, constant coalescent population size model using tuberculosis notification dates to calibrate tips (Supplementary Methods).
Transmission inference
M. tuberculosis phylogenetic trees represent patterns of evolutionary relatedness between the consensus bacterial genomes sampled from different individuals. Because most outbreaks are incompletely sampled (i.e. an M. tuberculosis sequence is not available for every case) and individuals may be infected with diverse populations of M. tuberculosis, phylogenies do not represent the underlying history of transmission.(19) We used TransPhylo(19) to infer transmission linkages—including unsampled hosts—that were consistent with the underlying timed phylogenies and transmission process (i.e. generation time and sampling intervals). To focus on densely sampled trees that would not be dominated by unsampled hosts, we sliced phylogenies at 2012 in order to generate subtrees with a most recent common ancestor of 2012 or later for transmission inference, including 122 subtrees each with 2 to 23 tips and comprising 56% of isolates (528/935).
By sampling from the posterior distribution of transmission trees, we estimated the posterior probability that each individual within a transmission tree was infected by each other individual in the tree. We summarized the posterior set of transmission trees in TransPhylo with who acquired infection from whom (WAIFW) matrices, W, where Wi,j is the posterior probability of transmission from individual j to individual i. Many transmission events are unobserved because of incomplete sampling; therefore, the sum of transmission probabilities to any individual is often less than one. We combined transmission trees with incarceration histories to quantify the proportion of observed transmission attributable to people with a history of incarceration. An individual’s location at the time of their tuberculosis notification may not reflect where transmission occurred. To understand where infector-recipient pairs were at the time transmission occurred, we integrated transmission trees with incarceration information (Fig. 4c). First, we used the transmission trees to infer the posterior distribution of infection times for each recipient i. We then partitioned the infector-recipient transmission probability Wi,j across the recipient’s infection time distribution, resulting in  , the probability of transmission from individual j to individual i while j has incarceration status v and i has incarceration status u. Incarceration status was trichotomized as incarcerated, formerly incarcerated, and no incarceration history.
, the probability of transmission from individual j to individual i while j has incarceration status v and i has incarceration status u. Incarceration status was trichotomized as incarcerated, formerly incarcerated, and no incarceration history.
We then summed pairwise, location-specific transmission probabilities over all individual subtrees with respect to the incarceration status of both infector j and recipient i. We defined the population wide transmission matrix, 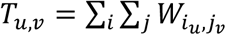 , where Tu,v is the sum of the posterior probabilities of observed transmission events to individuals in population u from individuals in population v. We normalized Tu,v by dividing by the sum of posterior transmission probabilities to each recipient population u, Nu = ∑v Tu,v. We then defined the uncorrected proportion of the total posterior transmission probabilities to population u from population v, or the transmission fraction, as
, where Tu,v is the sum of the posterior probabilities of observed transmission events to individuals in population u from individuals in population v. We normalized Tu,v by dividing by the sum of posterior transmission probabilities to each recipient population u, Nu = ∑v Tu,v. We then defined the uncorrected proportion of the total posterior transmission probabilities to population u from population v, or the transmission fraction, as  .
.
Genomic sampling varied across populations and specifically, we predicted that incarcerated people might have been sampled at a higher rate due to our active case finding and greater culture coverage in three prisons in the state. To correct for sampling bias, we determined the genomic sampling proportion rk, which we defined as the proportion of notified tuberculosis cases for which a high-quality genomic sequence was available, for population k at the time of tuberculosis notification. We calculated genomic sampling proportion as the number of sequenced genomes for each group divided by the number of tuberculosis notifications for each group from 2014-2019 in Campo Grande and Dourados, the two major cities in our prospective study (Fig. S2). We adjusted each pairwise transmission probability by the sampling rate of the infector, based on the incarceration status of the infector at the time of tuberculosis notification, for  and calculated the sampling-adjusted transmission fractions as described above.
and calculated the sampling-adjusted transmission fractions as described above.
Role of the funding source
The funding source had no role in study design, data collection, data analysis, data interpretation, or writing of the report.
Statistical methods
We inferred transmission trees with TransPhylo(19) as described above. We compared phylogenetic tree terminal branch lengths between populations with two-sample t-tests and compared the rate of clustering between populations with two-sample proportion tests.
Data Availability
Sequence data is available on the Sequence Read Archive (SRA), in BioProject PRJNA671770.
Funding
National Institutes of Health grant R01AI130058 (JRA)
Brazil’s National Council for Scientific and Technological Development grant 404237/2012-6 (JC)
Author contributions
Conceptualization: JRA, JC, KSW, CC, TC, BM, AIK
Methodology: JRA, JC, KSW, CC, TC, BM, AIK
Investigation: PCPS, TOG, BOS, ASS, ACL, AMS, FMFM, RDO, EFL, EC, YL
Visualization: KSW, JRA
Funding acquisition: JRA, JC
Project administration: JRA, JC
Supervision: JRA, JC
Writing – original draft: KSW, JRA
Writing – review & editing: all authors
Competing interests
Authors declare that they have no competing interests.
Data and materials availability
Raw Illumina sequence data are available on the Sequence Read Archive under BioProject PRJNA671770.
Acknowledgments
We would like to acknowledge the contributions of AGEPEN, LACEN, and the Mato Grosso do Sul State Health Department. All authors had full access to all the data in the study and had final responsibility for the decision to submit




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}