
ABSTRACT
Humans and viruses have co-evolved for millennia resulting in genetic polymorphisms that affect response to viral infection. We conducted a comprehensive study in the UK biobank linking germline genetic variation and gene expression with 28 antigens for 16 viruses in 7924 subjects. We discovered 7 novel loci associated with antibody response (P<5.0×10−8), including FUT2 for human polyomavirus BKV, TMEM173 for Merkel cell polyomavirus (MCV), and TBKBP1 for human herpesvirus 7. Transcriptome-wide analyses identified 114 genes association with response to viral infection, including ECSCR: P=5.0×10−15 (MCV), NTN5: P=1.1×10−9 (BKV), and P2RY13: P=1.1×10−8 (Epstein-Barr virus nuclear antigen). Signals in human leukocyte antigen (HLA) class II region dominated the landscape of viral antibody response, with 40 independent loci and 14 independent classical alleles, 7 of which exhibited pleiotropic effects across viral families. We also link viral response genes with complex diseases, such as C4A expression in varicella zoster virus and schizophrenia. Lastly, based on 1028 subjects tested for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), we identify 7 class II HLA susceptibility alleles (5 associated with other viruses). We also observe that genetic determinants of ACE2 expression may influence SARS-CoV-2 susceptibility. Our findings elucidate the genetic architecture of host response to viral infection, with potential implications for complex diseases and COVID-19.
INTRODUCTION
Viruses have been infecting cells for a half a billion years1. During our extensive co-evolution viruses have exerted significant selective pressure on humans; overtly during fatal outbreaks, and covertly through cryptic immune interaction when a pathogen remains latent. The recent pandemic of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) highlights the paramount public health need to understand human genetic variation in response to viral challenge. Clinical variation in COVID-19 severity and symptomatic presentation may be due to differences host genetic factors relating to immune response2. Furthermore, many common infections are cryptically associated with a variety of complex illnesses, especially those with an immunologic component, from cancer to autoimmune and neurologic conditions3–5. Despite their broad health relevance, few large scale GWAS studies have been conducted on viral infections6–10. Understanding the genetic architecture of immunologic response to viruses may therefore provide new insight into etiologic mechanisms of diverse complex diseases.
Some common viruses exert a robust cell mediated and humoral immune response that bi-directionally modulate the balance between latent and lytic infection. Previous studies have identified associations between host polymorphisms in genes relating to cell entry, cytokine production, and immune response and a variety of viruses11. Other studies have demonstrated a strong heritable component (32-48%) of antibody response12. Previous investigations have implicated human leucocyte antigen (HLA) class I and II in the modulation of immune response to diverse viral antigens7, 13. Furthermore, during the 2003 severe acute respiratory SARS-S outbreak, caused by a betacoronavirus related to SARS-CoV-2, an association with infection severity was reported for the HLA-B*46:01 allele in East Asian patients14.
In this study we utilize data from the UK Biobank (UKB) cohort15 to evaluate the relationship between host genetics and antibody response to 28 antigens for 16 viruses that have been linked to cancer and neurodegenerative diseases16, as well as SARS-CoV-2 test results in a separate subset of UKB participants. We conduct integrative genome-wide and transcriptome-wide analyses of antibody response and positivity to viral antigens, which elucidate novel genetic underpinnings of viral infection and immune response.
RESULTS
A random sample of the participants representative of the full UKB cohort was assayed using a multiplex serology panel16. We analyzed data from 7924 participants of predominantly European ancestry, described in Supplementary Table 1. Approximately 90% of individuals were seropositive for herpes family viruses with ubiquitous exposure: Epstein-Barr virus (EBV EA-D: 86.2% to ZEBRA: 91.2%), Human Herpesvirus 7 (HHV-7 94.8%), and Varicella Zoster Virus (VZV 92.3%). Seroprevalence was somewhat lower for cytomegalovirus (CMV), ranging between 56.5% (CMV pp28) and 63.3% (CMV pp52), and Herpes Simplex virus-1 (HSV1 69.3%). Human polyomavirus BKV was more prevalent (95.3%) compared to other polyomaviruses, Merkel cell polyoma virus (MCV 66.1%) and JCV (56.6%). Less common infections included HSV-2 (15.2%), HPV16 (E6 and E7 oncoproteins: 4.7%), HPV18 (2.4%), HTLV1 (1.6%), Hepatitis B (HBV, 1.6%), and Hepatitis C (HCV, 0.3%).
Baseline characteristics of 1474 UKB participants with SARS-CoV-2 test results up to April 14, 2020 are described in Supplementary Table 2. Compared to the full UKB cohort, individuals who were tested for SARS-CoV-2 were more likely to be smokers (55.1% vs. 45.2%), had higher mean BMI (28.7 vs. 27.4 kg/m2), higher levels of deprivation based on the Townsend index, and a higher burden of comorbidities based on the Charlson index (1 or more: 50.0% vs. 29.8%). Individuals with at least one positive test result (n=669) included a higher proportion of men (56.5%), former smokers (43.2% vs. 38.3%), and self-identified as non-white (15.5% vs. 8%). The two groups had similar distributions of other health-related characteristics. The final analytic dataset included 443 cases and 585 controls of European ancestry with test results from respiratory samples, excluding results from serum or unknown origins.
Genetic Determinants of Response to Viral Infection
Results from our GWAS of antibody response phenotypes were dominated by signals in the HLA region, which were detected for all EBV antigens (EA-D, EBNA, p18, ZEBRA), CMV pp52, HSV1, HHV7, VZV, JCV and MCV (Table 1; Supplementary Figure 1). Most of the top-ranking HLA variants for each antigen were independent of those for other antigens (Supplementary Figure 2). Exceptions were moderate LD between lead variants for EBV ZEBRA and HSV1 (r2=0.45), EBV EBNA and JCV (r2=0.45), and HHV7 and MCV (r2=0.44). Outside of the HLA region, genome-wide significant associations with seroreactivity were detected for: MCV at 3p24.3 (rs776170649, LOC339862: P=1.7×10−8) and 5q31.2 (rs7444313, TMEM173: P=2.4×10−15); BKV at 19q13.3 (rs681343, FUT2: P=4.7×10−15) (Figure 1); EBV EBNA at 3q25.1 (rs67886110, MED12L: P=1.3×10−9); HHV-7 at 11q23.3 (rs75438046, CXCR5: P=1.3×10−8) and 17q21.3 (rs1808192, TBKBP1: P=9.8×10−9); and HSV-1 at 10q23.3 (rs67886110: P=1.3×10−9).
Lead genome-wide significant variants (P<5.0×10−8) for continuous antibody response phenotypes for antigens with at least 20% seroprevalence.
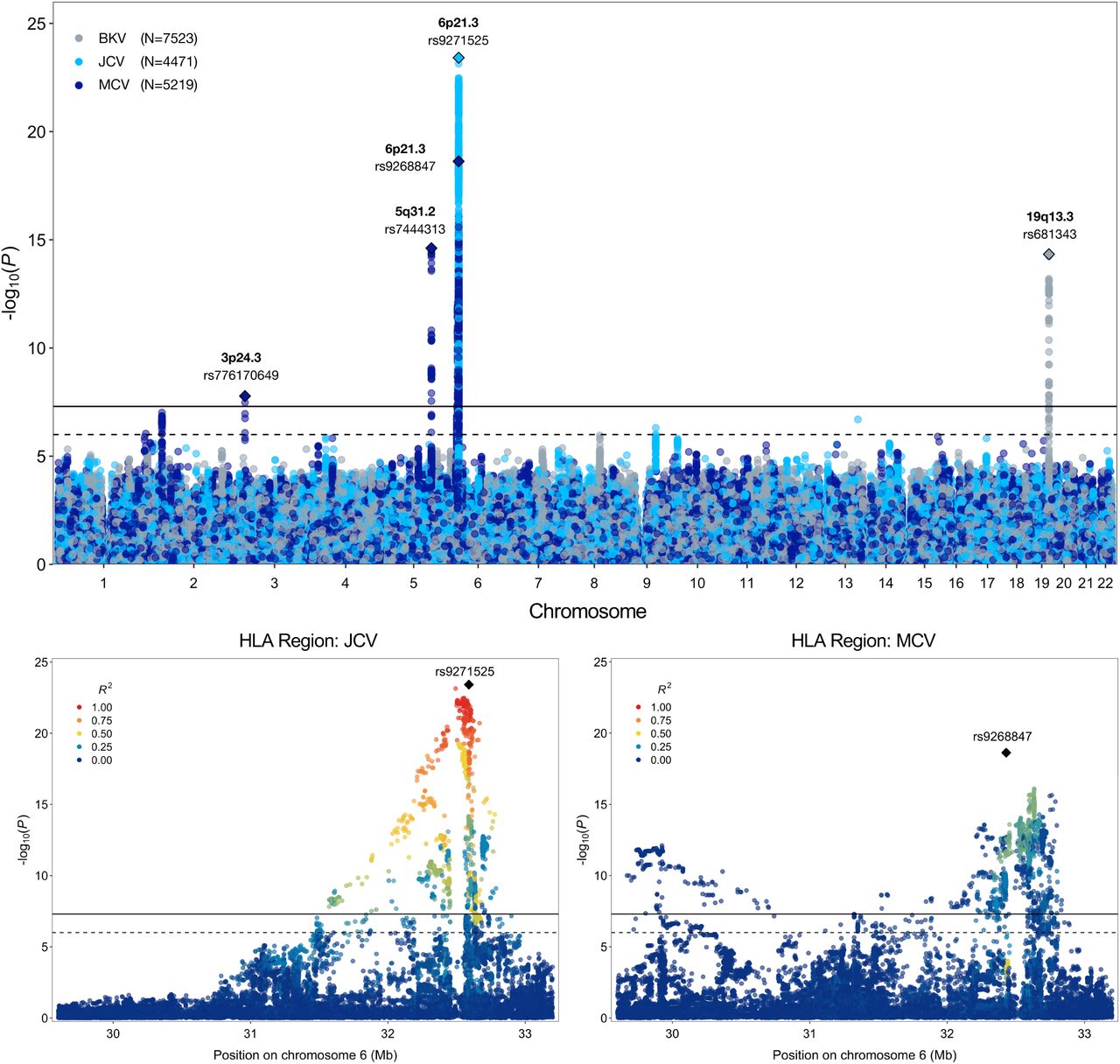
Results from genome-wide and regional association analyses of continuous antibody response phenotypes (MFI z-scores) among individuals seropositive for human polyomaviruses BKV, JCV, and Merkel cell (MCV). The lower two panels depict the association signal and linkage disequilibrium (LD) structure in the HLA region for JCV and MCV.
GWAS of discrete seropositivity phenotypes identified associations in HLA for EBV EA-D (rs2395192: OR=0.066, P=4.0×10−19), EBV EBNA (rs9268848: OR=1.60, P=1.2×10−18), EBV ZEBRA (rs17211342: 0.63, P=1.6×10−15), VZV (rs3096688: OR=0.70, P=3.7×10−8), JCV (rs9271147: OR=0.54, P=1.3×10−42), and MCV (rs17613347: OR=0.61, P=1.2×10−26) (Supplementary Figure 1; Supplementary Table 3). An association with susceptibility to MCV infection was also observed at 5q31.2 (rs1193730215, ECSCR: OR=1.26, P=7.2×10−9), with high LD (r2=0.95) between the lead variants for the seroreactivity and seropositivity phenotypes.
Several significant associations were observed for antigens with <20% seroprevalence, which were not included in the GWAS of antibody response due to inadequate sample size (Supplementary Table 3). Significant associations with infection susceptibility were observed for HSV2 in 17p13.2 (rs2116443: odds ratio (OR)=1.28, P=4.5×10−8; ITGAE); HPV16 E6 and E7 oncoproteins in 6p21.32 (rs601148: OR=0.60, P=3.3×10−9; HLA-DRB1) and 19q12 (rs144341759: OR=0.383, P=4.0×10−8; CTC-448F2.6); and HPV18 in 14q24.3 (rs4243652: OR=3.13, P=7.0×10−10). Associations were also detected for KSHV, HTLV1, HBV and HCV, including a variant in the MERTK oncogene (HCV Core rs199913364: OR=0.25, P=1.2×10−8).
Functional Characterization of GWAS Findings
In-silico functional analyses of the lead 17 GWAS variants identified enrichment for multiple regulatory elements (summarized in Supplementary Table 4). Three variants were predicted to be in the top 10% of deleterious substitutions in GRCh37 based on CADD scores >10: rs776170649 (MCV, CADD=15.61), rs139299944 (HHV7, CADD=12.15), and rs9271525 (JCV, CADD=10.73). Another HHV7-associated variant, rs1808192 (RegulomeDB rank: 1f), an eQTL and sQTL for TBKBP1, mapped to 44 functional elements for multiple transcription factors (TF), including IKZF1, a critical regulator of lymphoid differentiation frequently mutated in B-cell malignancies.
Eleven sentinel variants were eQTLs and 8 were splicing QTLs in GTEx, with significant (FDR<0.05) effects across multiple genes and tissues (Supplementary Figure 3). The most common eQTL and sQTL targets included HLA-DQA1, HLA-DQA2, HLA-DQB1, HLA-DQB2, HLA-DRB1, and HLA-DRB6. Outside of HLA, rs681343 (BKV), a synonymous FUT2 variant was an eQTL for 8 genes, including FUT2 and NTN5. MCV variant in 5q31.2, rs7444313, was an eQTL for 7 genes, with concurrent sQTL effects on TMEM173, also known as STING1 (stimulator of interferon response cGAMP interactor 1) and CXXC5. Gene expression profiles in immune cell populations from DICE17 identified several cell-type specific effects that were not observed in GTEx. An association with HLA-DQB1 expression in CD4+ TH2 cells was observed for rs9273325, 6:31486158_GT_G was an eQTL for ATP6V1G2 in naïve CD4+ T cells, and rs1130420 influenced the expression of 8 HLA class II genes in naïve B-cells and CD4+ TH17 cells.
We identified 7 significant (p<5.0×10−8) protein quantitative trait loci (pQTL) for 38 proteins (Supplementary Table 5). Most of the pQTL targets were components of the adaptive immune response, such as the complement system (C4, CFB), chemokines (CCL15, CCL25), and defensin processing (Beta-defensin 19, Trypsin-3). The greatest number and diversity of pQTL targets (n=16) was observed for rs681343, including BPIFB1, which plays a role in antimicrobial response in oral and nasal mucosa18; FUT3, which catalyzes the last step of Lewis antigen biosynthesis; and FGF19, part of the PI3K/Akt/MAPK signaling cascade that is dysregulated in cancer and neurodegenerative diseases19.
Pleiotropic associations with disease outcomes
To contextualize the relevance of genetic loci involved in infection response, we explored associations with selected cancers, schizophrenia, and that have a known or suspected viral etiology (Supplementary Table 6). The strongest secondary signal was observed for rs9273325, which was negatively associated with VZV antibody response and positively associated with schizophrenia susceptibility (OR=1.13, P=4.3×10−15). Other significant (Bonferroni P<7.4×10−4) associations with schizophrenia were detected for HSV1 (rs1130420: OR=1.06, P=1.8×10−5), EBV EA-D (rs2647006: OR=0.96, P=2.7×10−4), BKV, (rs681343: OR=0.96, P=2.5×10−4) and JCV (rs9271525: OR=1.06, P=6.8×10−5). Inverse associations with hematologic cancers were observed for HSV1 (rs1130420: OR=0.89, P=3.5×10−6), VZV (rs9273325: OR=0.88, P=4.4×10−5), and EBV EBNA (rs9269233: OR=0.88, P=2.7×10−4) variants. HSV1 antibody response was also linked to Alzheimer’s disease (rs1130420: P=1.2×10−4).
Regional HLA Associations
Associations within the HLA region were refined by identifying independent (LD r2<0.05 within ±500kb) sentinel variants with P<5.0×10−8 for each antigen response phenotype (Supplementary Table 7). Clumping seropositivity associations with respect to lead antibody response variants did not retain any loci, suggesting non-independence in signals for infection and reactivity for the same antigen. For this reason, all subsequent analyses focus on seroreactivity phenotypes. To assess the independence of HLA associations for different antigens, we performed clumping across phenotypes, which identified 40 independent (LD r2<0.05 within ±500kb) sentinel variants: EBV EBNA (12), VZV (11), EBV ZEBRA (8), EBV p18 (5), MCV (3), and EBV EA-D (1) (Supplementary Table 9). No LD clumps were anchored by variants detected for CMV pp52, HHV7, HSV1, or JCV, suggesting that the HLA signals for these antigens are captured by lead loci for other phenotypes. The largest region with the lowest p-value was anchored by rs9274728 (P=4.7×10−67) near HLA-DQB1, originally detected for EBV ZEBRA. Of the 11 VZV-associated variants, the largest clump was formed around rs4990036 (P=4.5×10−26) in HLA-B.
Iterative conditional analyses adjusting for the HLA SNP/indel with the lowest p-value were performed until no variants remained with Pcond<5.0×10−8. Additional independent variants were identified for EBV EBNA (rs139299944, rs6457711, rs9273358, rs28414666, rs3097671), EBV ZEBRA (rs2904758, rs35683320, rs1383258), EBV p18 (rs6917363, rs9271325, rs66479476), and MCV (rs148584120, rs4148874) (Figure 2; Supplementary Table 8). For CMV pp52, HHV7, HSV1, JCV, and VZV, the regional HLA signal was captured by the top GWAS variant (Figure 2; Supplementary Table 8).

Regional association plots for conditionally independent HLA genetic variants that were significantly (P<5.0×10−8, solid black line) associated with each continuous antibody response phenotype. The suggestive significance threshold corresponds to P<1.0×10−6 (dotted black line).
Next, we tested 101 classical HLA alleles and performed analogous iterative conditional analyses. Significant associations across viruses were predominantly observed for class II HLA alleles. Five statistically independent signals were identified for antibody response to EBV ZEBRA (DRB4*99:01: P=1.4×10−46; DQB1*04:02: Pcond=1.0×10−19; DRB1*04:04: Pcond=1.1×10−18; DQA1*02:01: Pcond=1.1×10−10, A*03:01: Pcond=1.9×10−8) and EBV EBNA (DRB5*99:01: P=8.7×10−30; DRB3*02:02: Pcond=6.8×10−30; DQB1*02:01: Pcond=3.6×10−12; DRB4*99:01: Pcond=8.3×10−17; DPB1*03:01: Pcond=4.7×10−14) (Figure 3; Supplementary Tables 10-11). Fewer independent alleles were observed for EBV p18 (DRB5*99:01: P=1.7×10−22; DRB1*04:04: Pcond=1.3×10−18) (Figure 3; Supplementary Tables 12).

Conditionally independent classical HLA alleles significantly (Pcond<5.0×10−8, solid red line) associated with each continuous antibody response phenotype. Only classical alleles that surpassed the Bonferroni-corrected significance threshold (P<5.0×10−4, dotted red line) were included in conditional analyses.
DQB1*02:01 was the only independently associated allele for EBV EA-D (β=-0.154, P=8.4×10−11) and HSV1 (β=0.145, P=2.8×10−8), although its effects were in opposite directions for each antigen (Supplementary Table 13). For VZV, associations with 16 classical alleles were accounted for by DRB1*03:01 (P=7.3×10−26). JCV shared the same lead allele as EBV EBNA and EBV p18 (DRB5*99:01: P=1.2×10−21) (Supplementary Table 13). Four conditionally independent signals were identified for MCV (DQA1*01:01: P=1.1×10−15; DRB1*04:04: Pcond=3.0×10−11; A*29:02: P=1.0×10−11; DRB1*15:01: P=3.7×10−12) (Figure 3; Supplementary Table 14).
Lastly, we integrated associations across variant types by including conditionally independent HLA alleles as covariates in the SNP-based analysis. With the exception of EBV antigens and HHV7, the classical alleles captured all genome-wide significant SNP signals (Supplementary Figure 5).
Genetic Associations With SARS-CoV-2 Status
We detected a suggestive association between rs7231584 in LDLRAD4, a low-density lipoprotein receptor gene, and having a positive SARS-CoV-2 test (OR=1.66, P=1.6×10−7) (Table 2; Supplementary Table 4). This variant has high regulatory potential (RegulomeDB probability score: 0.88), is an eQTL for RP11-,53B2.2, and overlaps with TF binding motifs for SP1, which is implicated in HSV1 infection20.
Genetic variants associated with laboratory-confirmed SARS-Cov-2 in a subset of UK Biobank participants with available test results. Odds ratios (OR) for testing positive were estimated in 1028 individuals of predominantly European ancestry excluding test results based on serum, skin swabs, and specimens of unknown origins.
Seven HLA alleles were nominally associated with testing positive for SARS-CoV-2 (P<0.05), five of which were also associated with antibody response to multiple viral antigens at the Bonferroni-corrected threshold for 101 alleles (P<5×10−4) (Table 2). DQA1*01:02 was positively associated with SARS-CoV-2 (OR=1.39, 1.08-1.77) and negatively associated with antibody response to 7 antigens (EBV p18, EBV ZEBRA, HHV7, HSV1, VZV, JCV, and MCV). DRB1*15:01 (OR=1.33, 1.01-1.74). DQB1*06:02 also increased the likelihood of testing positive for SARS-CoV-2 (OR=1.32, 1.00-1.74). This allele was inversely associated with response to EBV ZEBRA, HSV1, VZV, JCV, MCV, and positively associated with EBV EBNA and p18 response. Two HLA alleles were inversely associated with SARS-CoV-2 infection: DPB1*10:01 (OR=0.31, 0.12 - 0.76) and DQB1*05:01 (OR=0.75, 0.57-1.00), the latter being the top HHV7-associated allele.
ACE2 mediates SARS-CoV-2 cell entry21, therefore we investigated the potential associations between eQTLs for this gene and testing positive for SARS-CoV-2. We identified 288 significant (qFDR<0.05) eQTLs for ACE2 in GTEx v8 and estimated each variant’s association with a positive SARS-CoV-2 test result. Four variants in LD (r2>0.90) were nominally associated with SARS-CoV-2 status (P<0.05), led by rs1567893 (OR=1.18, 1.01-1.39) (Table 2). Considering only independent (LD r2<0.10) eQTLs, we observed a negative correlation between ACE2 expression and testing positive for SARS-CoV-2 (−0.367, P=0.034) (Supplementary Figure 6).
TWAS of Genes Involved in Antibody Response
Based on known targets of infection or related pathologies, we considered expression in the frontal cortex (Supplementary Table 15), EBV-transformed lymphocytes for EBV antigens (Supplementary Table 16), and skin for MCV (Supplementary Table 17). Concordance across tissues was summarized using Venn diagrams (Figure 4; Supplementary Figure 5). TWAS identified 114 genes significantly associated (PTWAS<4.2×10−6) with antibody response in at least one tissue, 54 of which were associated with a single phenotype, while 60 influenced seroreactivity to multiple antigens. We also include results for 92 additional suggestively (PTWES<4.5×10−5) associated genes.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
TWAS associations with continuous antigen response phenotypes. Two Manhattan plots depicting the transcriptome-wide associations for genes with a positive direction of effect (increased expression leads to higher antibody response) and genes with a negative direction of effect (increased expression is associated with a reduced antibody response).
The TWAS results mirrored earlier findings, with a predominance of associations in HLA class II genes. Some of the strongest overall associations were observed for HLA-DRB5 (EBV ZEBRA: Pcortex=4.2×10−45) and HLA-DRB1 (EBV EBNA: Pcortex=6.7×10−39; EBV ZEBRA: Pcortex=3.3×10−33; JCV: Pcortex=6.5×10−14; EBV p18: Pcortex=2.2×10−12). Increased expression of HLA-DQB2 was positively associated with antibody response to EBV ZEBRA (Pblood=7.6×10−19), JCV (Pblood=9.9×10−10), VZV (Pblood=7.0×10−9), HHV7 (Pblood=7.3×10−8), and HSV1 (Pblood=3.3×10−7), but negatively associated with EBV EBNA (Pblood=3.6×10−34) and EBV p18 (Pblood=2.1×10−8), in a consistent manner across tissues. The opposite was observed for HLA-DQB1, with positive effects on EBV EBNA and EBV p18 and inverse associations with EBV ZEBRA, JCV, VZV, HHV7, and HSV1.
The TWAS analyses also identified a number of significant associations in the HLA class III region that were not detected in other analyses. The top-ranking VZV associated gene was APOM (Pblood=7.5×10−27, Pcortex=1.1×10−25). Interestingly, opposite directions of effect for C4A and C4B gene expression. Increased C4A expression was positively associated with all EBV antigens (Supplementary Table 16), but negatively associated with VZV (Pblood=2.3×10−24) and HSV1 (Pcortex=1.8×10−5) antibody levels (Supplementary Table 15). On the other hand, increased C4B expression was inversely associated with EBV phenotypes, but positively associated with VZV (Pblood=8.1×10−25) and HSV1 (Pblood=1.1×10−5). A similar pattern was also observed for CYP21A2 and C2, with positive effects on antibody response to VZV and HSV1, and negative effects for all EBV antigens. Other novel TWAS findings were detected for HHV7 in 22q13.2 (CTA-223H9.9: PTWAS=2.5×10−6; CSDC2: PTWAS=3.0×10−6; TEF: PTWAS=3.1×10−6) and 1q31.2 (RGS1: PTWAS=3.3×10−6).
The TWAS recapitulated several GWAS-identified loci: 3q25.1 for EBV EBNA (P2RY13: Pcortex=1.1×10−8; P2RY12: Pblood=3.3×10−8) and 19q13.33 for BKV (FUT2: PTWAS=8.1×10−13; NTN5: PTWAS=1.1×10−9). Transcriptomic profiles in skin tissues provided supporting evidence for the role of multiple genes in 5q31.2 in modulating MCV antibody response (Figure 4; Supplementary Table 17). The strongest signal was observed in for ECSCR (skin sun unexposed: PTWAS=5.0×10−15; skin sun exposed: PTWAS=4.2×10−13), followed by PROB1 (sun unexposed: PTWAS=1.5×10−11). ECSCR expression was also associated based on expression in the frontal cortex, while PROB1 exhibited a significant, but attenuated effect in whole blood. VWA7 was the only gene associated across all four tissues for MCV and was also associated with antibody response to several EBV antigens.
Comparison of results for seroreactivity and seropositivity revealed a number of genes implicated in both steps of the infection process (Supplementary Table 18). Associations with HLA DQA and DQB genes in whole blood and HLA-DRB genes in the frontal cortex were observed for EBV antigens, JCV, and MCV. For MCV, the strongest seropositivity signals were observed for HLA class III genes AGER (Pcortex=9.0×10−21) and EHMT2 (Pblood=5.8×10−18), which were also among the top-ranking genes for seroreactivity. Increased ECSCR expression conferred an increased susceptibility to MCV infection (Pcortex=1.8×10−8), mirroring its effect on seroreactivity. In contrast to antibody response, no significant associations with any HLA genes were observed for VZV seropositivity.
Analyses using the Reactome database identified significant (qFDR<0.05) enrichment for TWAS-identified genes in pathways involved in initiating antiviral responses, such as MHC class II antigen presentation, TCR signaling, and interferon (IFN) signaling (Supplementary Figure 7). Pathways unique to herpes viruses included folding, assembly and peptide loading of class I MHC (q=3.2×10−7) and initial triggering of complement (q=9.8×10−3). Polyomaviruses were associated with the non-canonical nuclear factor (NF)-κB pathway activated by tumor necrosis factor (TNF) superfamily (q=1.9×10−3). Protein interaction networks among genes associated with more than antigen and identified 21 significant (qFDR<0.05) nodes, most of which were centered around HLA-DRB1 and HLA-DQB1, and involved interactions with other MHC class II molecules, C4, and TNF (Supplementary Figure 8).
DISCUSSION
We performed genome-wide and transcriptome-wide association studies for serological phenotypes for 16 common viruses in a well-characterized, population-based cohort. Our findings confirm that HLA class II and III genes are crucial host genetic factors involved in regulating immune response to diverse viral antigens7. We discover that specific HLA alleles that are associated with multiple common infections are also associated with SARS-CoV-2 positive tests. Furthermore, we uncovered novel genetic determinants of viral antibody response beyond the HLA region for BKV, MCV, HHV7, and EBV EBNA. This analysis provides a resource for further understanding the complex interplay between viruses and the human genome, as well as a first step towards understanding germline determinants of SARS-CoV-2 infection.
One of our main findings is the discovery of 5q31.2 as a susceptibility locus for MCV infection and MCV antibody response, implicating two main genes: TMEM173 (or STING1) and ECSCR. The former encodes STING (stimulator of interferon genes), an endoplasmic reticulum (ER) protein that controls the transcription of host defense genes and plays a critical role in response to DNA and RNA viruses22. STING is activated by cyclic GMP-AMP synthase (cGAS), a cytosolic DNA sensor that mounts a response to invading pathogens by inducing IFN1 and NF-κB signalling23,24. Polyomaviruses penetrate the ER membrane during cell entry, a process that may be unique to this viral family25, which may trigger STING signaling in a distinct manner from other viruses25. Multiple cancer-causing viruses, such as KSHV, HBV, and HPV18, encode oncoproteins that disrupt cGAS-STING activity, which illustrates the evolutionary pressure on DNA tumor viruses to develop functions against this pathway and its importance in carcinogenesis23. Furthermore, cGAS-STING activation has been shown to trigger antitumor T-cell responses, a mechanism that can be leveraged by targeted immunotherapies 26–28. Several studies suggest STING agonists may be effective against tumors resistant to PD-1 blockade, as well as promising adjuvants in cancer vaccines29–31.
ECSCR expression in skin and brain tissues was associated with MCV antibody response and infection. This gene encodes an endothelial cell-specific chemotaxis regulator, which plays a role in angiogenesis and apoptosis32. ECSCR is a negative regulator of PI3K/Akt signaling by enhancing membrane localization of PTEN and operates in tandem with VEGFR-2 and other receptor tyrosine kinases33. In addition to 5q31.2, another novel MCV seroreactivity associated region was identified in 3p24.3, anchored by rs776170649, which has been linked to platelet phenotypes34. These findings align with an increasingly recognized role of platelet activation in defense against infections via degranulation and release of chemokines and β-defensin35.
Genetic variation within Fucosyltransferase 2 (FUT2) has been studied extensively in the context of human infections; however, its effect on BKV seroreactivity is novel. Homozygotes for the nonsense mutation (rs601338 G>A) that inactivates the FUT2 enzyme are unable to secrete ABO(H) histo-blood group antigens or express them on mucosal surfaces36,37. The allele which confers increased BKV antibody response (rs681343-T) is in LD (r2=1.00) with rs601338-A, the non-secretor allele, which confers resistance to norovirus38,39, rotavirus40, H. pylori41, childhood ear infection, mumps, and common colds13. However, increased susceptibility to other pathogens, such as meningococcus and pneumococcus42 has also been observed in non-secretors. Isolating the underlying mechanisms for BKV response is challenging because FUT2 is a pleiotropic locus associated with diverse phenotypes, including autoimmune and inflammatory conditions43,44, serum lipids45, B vitamins37,46, alcohol consumption47, and even certain cancers48. In addition to FUT2 in 19q 13.33, NTN5 (netrin 5) suggests a possible link between BKV and neurological conditions. NTN5 is primarily expressed in neuroproliferative areas, suggesting a role in adult neurogenesis, which is dysregulated in glioblastoma and Alzheimer’s disease49,50.
We also report the first GWAS of serological phenotypes for HHV7. Genetic determinants of HHV7 antibody response in 6p21.32 were predominantly localized in HLA-DQA1 and HLA-DQB1, with associations similar to other herpes viruses. In 11q23.3, rs75438046 maps to the 3’ UTR of CXCR5, which controls viral infection in B-cell follicles51, and BCL9L, a translocation target in acute lymphoblastic leukemia52 and transcriptional activator of the Wnt/β-catenin cancer signaling pathway53. In 17q21.32, TBKBP1 encodes an adaptor protein part of the TNF/NF-κB interaction network, where it regulates immune responses to infectious triggers, such as IFN1 signaling54. Consistent with diverse functions of this pathway in cell survival and proliferation, the TBK1-TBKBP1 axis also mediates tumor growth and immunosuppression through induction of PD-L155.
Several additional genes involved in HHV7 immune response were identified in TWAS. TEF in 22q13.2 is an apoptotic regulator of hematopoietic progenitors with tumor promoting effects mediated by inhibition of G1/S cell cycle transition and Akt/FOXO signaling56. RGS1 in 1q31.2 has been linked to multiple autoimmune diseases, including multiple sclerosis57, as well as poor prognosis in melanoma and diffuse large B cell lymphoma mediated by inactivation of Akt/ERK58,59.
The last novel locus associated with viral infection response outside of the HLA region was 3q25.1, detected for EBV EBNA. The lead variant (rs67886110) is an eQTL for MED12L and P2RY12 genes, which have been linked to neurodegenerative conditions60,61. P2RY12 and P2RY13, identified in TWAS, are purinergic receptor genes that regulate microglia homeostasis and have been implicated in Alzheimer’s susceptibility via inflammatory and neurotrophic mechanisms61.
Considering genetic variation within the HLA region, our results not only confirm its pivotal role at the interface of host pathogen interactions, but also highlight the overlap in variants, classical alleles, and genes that mediate these interactions across virus families and antigens. We identified 40 independent SNPs/indels associated with EBV (EBNA, EA-D, VCA p18, and ZEBRA), VZV, and MCV antibody response that accounted for all significant HLA associations for other phenotypes. Of the 14 conditionally independent, genome-wide significant classical alleles identified for 10 antigens, 7 were associated with multiple phenotypes. The most commonly shared HLA alleles were DRB5*99:01, DRB1*04:04, an know rheumatoid arthritis risk allele62, and DQB1*02:01, which is implicated in susceptibility to celiac disease63. Furthermore, nodes anchored by HLA-DRB1 and HLA-DQB1 were at the center of the protein interaction network identified for TWAS genes that were implicated in antibody response to multiple antigens. Despite the predominance of association in HLA class II, one notable association in HLA class I was detected for A*29:02 and MCV seroreactivity, which is consistent with downregulation of MHC I as a potential mechanism through which Merkel cell tumors evade immune surveillance64.
Comparison with other studies of host genetics and viral infection susceptibility shows that our results align with previously reported findings. We replicated (P<5×10−8) many associations from a GWAS of humoral immune response by Hammer et al.7, specifically HLA signals for MCV (rs1049130, DQB1*06:02), JCV (rs9269910, DQA1*01:02), and EBV EBNA (DRB1*07:01, HLA-DRB1*03:01, also reported by Scepanovic et al8). Associations with JCV seroreactivity replicated (P<5×10−8) class II HLA alleles (DQB1*03:01, DQB1*06:02, and DQA1*01:02) linked to JCV infection in a cohort of multiple sclerosis patients and population-based controls65. We also replicated two HLA-DRB1 variants (rs477515, rs2854275: P<5×10−19) associated with EBV EBNA antibody levels in a Mexican American population9. Our GWAS of HPV16 L1 replicated a variant previously linked to HPV8 seropositivity (rs9357152, P=0.008)6. Some of our findings contrast with Tian et al 13, although we confirmed selected associations, such as A*02:01 (shingles) with VZV (P=4.1×10−8) and rs2596465 (mononucleosis) with EBV EBNA (P=3.3×10−9) and EBV p18 (P=1.0×10−12). These differences may be partly accounted for by the use of self-reported disease status in Tian et al. which may be an imprecise indicator of infection with certain viruses.
One of the most striking findings in SNP-based HLA analyses was the genome-wide significant association between rs9273325, sentinel VZV antibody response variant, and risk of schizophrenia. This observation is consistent with the established role the HLA region, including HLA-DQB1, in schizophrenia etiology66,67, and is further supported by previously reported associations for rs9273325 with blood cell traits34 and immunoglobulin A deficiency68, as well as its role as an eQTL for HLA-DQB1 in CD4+ T2h cells. An inverse association with schizophrenia has been reported for DRB1*03:0166, the lead HLA allele associated with increased VZV antibody response. Two other strongly associated schizophrenia alleles, HLA-DQB1:02 and HLA-B*08:01, were also among the top VZV-associated variants (P<5×10−25) in the unconditional analysis. Enhanced complement activity has been proposed as the mechanism mediating the synaptic loss and excessive pruning which is a hallmark of schizophrenia pathophysiology69. Complement component 4 (C4) alleles were found to increase risk of schizophrenia proportionally to their effect on increasing C4A expression in brain tissue69. Using gene expression models in whole blood and the frontal cortex we demonstrated that increased C4A expression is negatively associated with VZV antibody response. We also observed associations with C4A and C4B in EBV and HSV-1, but not other viruses. Taken together, these findings delineate a potential mechanism through which aberrant immune response to VZV infection, and potentially HSV-1 and EBV infection, may increase susceptibility to schizophrenia.
Our finding that certain HLA alleles are associated with SARS-CoV-2 may shed light on the diverse clinical presentations of this infection, including asymptomatic cases and severe COVID-19 in younger patients and those with few pre-existing conditions70,71. Associations between SARS-CoV-2 status and HLA alleles may reflect a different clinical course with milder infection and/or asymptomatic status, which led to lower likelihood of being tested despite exposure. This is especially true since our data were collected early in the epidemic, when patients with more severe disease were prioritized for testing. If true, these associations may be stronger when comparing more severe disease COVID+ patients to asymptomatic COVID+ carriers. Although we could not directly test this hypothesis, future studies of symptom severity may clarify the clinical implications of these associations. Most of the SARS-CoV-2 associated HLA alleles displayed broad pleiotropic effects across multiple viral families, particularly risk-increasing DQA1*01:02, DRB1*15:01, and DQB1*06:02 alleles which comprise a common haplotype in European ancestry individuals that confers protection against type 1 diabetes72, yet increases risk of multiple sclerosis73. These alleles may affect immunity to multiple viral antigens differentially, resulting in varying efficiency recognizing viral epitopes and subsequent presentation to CD8+ T cells.
We identified two HLA alleles which may confer protection against SARS-CoV-2 infection: DPB1*10:01 and DQB1*05:01. Previous studies have linked DQB1*05:01 to Guillain-Barre syndrome, which can result from viral challenge or vaccination74 and recently Guillain-Barre has been clinically observed in some COVID-19 patients75. We were unable to assess the role of HLA-B*46:01, previously linked to 2003 SARS in an East Asian population, due to the low frequency (3.2×10−5) of this allele in our data. This illustrates the need for studies in large, genetically diverse populations to elucidate the role of HLA in SARS-CoV-2 infection dynamics.
We also demonstrate an inverse relationship between genetically predicted ACE2 gene expression and having a positive SARS-CoV-2 test. Although ACE2 plays a critical role in facilitating SARS-CoV-2 cell entry21, SARS-CoV-2 appears to down-regulate ACE2 expression upon establishment in the cell76. Another betacoronavirus SARS-S, has shown similarity in ACE2 expression demonstrating a protective effect on lung injury76,77. We suspect that decreased ACE2 expression may reduce viral infection by limiting available receptors for viral entry, and once a cell is infected, reduced expression may aid SARS-CoV-2. Further research is needed to understand this relationship and therapeutic research on ACE2 inhibitors will elucidate these mechanisms. The association of rs7231584 with SARS-CoV-2 status may also be promising due to the role of LDLRAD4 as a negative regulator of TGF-β signaling78.
While the reported associations for SARS-CoV-2 were based on a limited sample size, we believe these findings may be informative when considered in the context of our results for other viral infections, and contribute to the global, pressing need to understand COVID-19, which motivated our rapid analysis of these data. However care must be taken with the interpretation of genetic associations with SARS-CoV-2 status, which should be regarded as preliminary and exploratory given the small number of tests within a biased sample, confounding by factors related to symptom recognition, access to testing, and limited statistical power.
Several additional limitations of this work should be noted. First, the UK Biobank is unrepresentative of the general UK population due to low participation resulting in healthy volunteer bias79. However, since the observed pattern of seroprevalence is consistent with previously published estimates16 we believe the impact of this bias is likely to be minimal. Second, our analyses were restricted to participants of European ancestry due to limited serology data for other ancestries, which limits the generalizability of our findings to diverse populations. Third, we were unable to conduct formal statistical replication of novel GWAS and TWAS signals in an independent sample due to the lack of such a population. Nevertheless, our successful replication of multiple previously reported variants and, combined with the observation that newly discovered genes and variants are part of essential adaptive and innate immunity pathways, support the credibility of our findings. Lastly, we We also stress caution in the interpretation of GWAS results for non-ubiquitous pathogens, such as HBV, HCV, and HPV, due to a lack of information exposure to as well as low numbers of seropositive individuals.
Our study also has distinct advantages. The large sample size of the UK Biobank facilitated more powerful genetic association analyses than previous studies, in a population-based cohort unselected for disease status. Our detailed HLA analysis shows independent effects of specific HLA alleles, and pleiotropic effects across multiple viruses. Analyses of genetic associations in external datasets further demonstrate a connection between host genetic factors influencing immune response to infection and susceptibility to cancers and neurological conditions. Finally, integration of SARS-CoV-2 test results with findings for serology measures for common viruses provides previously unknown context for putative associations.
The results of this work highlight widespread genetic pleiotropy between pathways involved in regulating humoral immune response to novel and common viruses. We show associations with response to common viruses and complex diseases. Understanding the interplay between host genetic factors and immune response has implications for public health and may facilitate the discovery of novel therapeutics including vaccines. Upon further elucidation of the risk of infection and COVID-19 severity, our results suggest that HLA typing may be a feasible tool in the SARS-CoV-2 response for identifying at risk populations and, prioritizing vaccine distribution. Further research is required to understand the feasibility of this approach in diverse populations.
METHODS
Study Population and Phenotypes
The UK Biobank (UKB) is a population-based prospective cohort of over 500,000 individuals aged 40-69 years at enrollment in 2006-2010 who completed extensive questionnaires, physical assessments, and provided blood samples15. Analyses were restricted to individuals of predominantly European ancestry based on self-report and after excluding samples with any of the first two genetic ancestry principal components (PCs) outside of 5 standard deviations (SD) of the population mean. We removed samples with discordant self-reported and genetic sex, samples with call rates <97% or heterozygosity >5 SD from the mean, and a one sample from each pair of first-degree relatives identified using KING80.
Of the 413,810 European ancestry individuals available for analysis, a total of 7948 had serological measures. A multiplex serology panel was performed over a 2-week period using previously developed methods81,82 that have been successfully applied in epidemiological studies7,83. Details of the serology methods and assay validation performance are described in Mentzer et al.16 Briefly, multiple serology was performed using a bead-based glutathione S-transferase (GST) capture assay with glutathione-casein coated fluorescence-labelled polystyrene beads and pathogen-specific GST-X-tag fusion proteins as antigens16. Each antigen was loaded onto a distinct bead set and the beads were simultaneously presented to primary serum antibodies at serum dilution 1:100016. Formed immunocomplexes were quantified using a Luminex 200 flow cytometer, which produced Median Fluorescence Intensities (MFI) for each antigen. The serology assay showed adequate performance, with a median coefficient of variation (CV) of 17% across all antigens and maximum of 35.4% for H.pylori GroEL, which was excluded from our analysis16. Among seropositive samples only, the overall median CV was 3.5%16. The highest CV’s for antigens included in our study were observed for HHV6 IE1A (26.2% overall) and BKV (7.6% in seropositive only). Repeated measures in a 3-5-year interval on a subset of 277 participants suggested that seropositivity was relatively stable over time, with seroconversion rates between 0% and 7.9% (HHV6 IE1A/IE1B)16.
A total of 2724 records for SARS-CoV-2 test results between March 16, 2020 and April 14, 2020 was provided by Public Health England for 1474 UK Biobank participants. Test results based on serum, skin swabs, or specimens of unknown origin (n=318) were excluded due to low viral content and potentially unreliable detection2. Using the remaining 2406 records for 1369 individuals, COVID-19 cases were classified as individuals with any positive test based on respiratory specimens. After restricting to individuals of predominantly European ancestry 443 cases with at least one positive SARS-CoV-2 test result and 585 controls with negative SARS-CoV-2 test results remained.
Genome-Wide Association Analysis
We evaluated the relationship between constitutive genetic variants across the genome and antigens or SARS-CoV-2 status using PLINK 2.0 (October 2017 version). Participants were genotyped on the UK Biobank Affymetrix Axiom array (89%) or the UK BiLEVE array (11%)15 with genome-wide imputation performed using the Haplotype Reference Consortium data and the merged UK10K and 1000 Genomes (1000G) phase 3 reference panels15. We excluded variants out of Hardy-Weinberg equilibrium at p<1×10−5, call rate <95% (alternate allele dosage required to be within 0.1 of the nearest hard call to be nonmissing), imputation quality INFO<0.30, and MAF<0.01.
Seropositivity for each antigen was determined using established cut-offs based on prior validation work16. The primary antigen GWAS focused on those with seroprevalence of ≥20%, and investigated continuous phenotypes (MFI values), corresponding to the magnitude of antibody response or seroreactivity among seropositive individuals. MFI values were transformed to standard normal distributions using ordered quantile normalization84.
The antigen GWAS entailed linear regression of MFI z-scores on genotype. These regression models adjusted for age at enrollment, sex, body-mass index (BMI), socioeconomic status (Townsend deprivation index), the presence of any autoimmune and/or inflammatory conditions, genotyping array, serology assay date, quality control flag indicating sample spillover or an extra freeze/thaw cycle, and the top 10 genetic ancestry principal components (PC’s). Autoimmune and chronic inflammatory conditions were identified using the following primary and secondary diagnostic ICD-10 codes based on Hospital Episode Statistics: immune and infection-related arthropathies (M00-M03, M05-M14); multiple sclerosis (G35); Crohn’s disease, colitis, irritable bowel syndrome (K50-K52, K58); lupus (M32, L93); dermatitis, eczema, and psoriasis (L20-30, L40); inflammatory polyneuropathy (G61). Individuals diagnosed with any immunodeficiency (ICD-10 D80-89, n=24) were excluded from all analyses. For all antigens with at least 100 seropositive (or seronegative for pathogens with ubiquitous exposure) individuals, GWAS of discrete seropositivity phenotypes was undertaken using logistic regression, adjusting for the same covariates listed above.
The functional relevance of the lead GWAS loci for antibody response was assessed using in-silico analyses using integrative summary scores, Combined Annotation Dependent Depletion (CADD)85 and RegulomeDB 2.086, and by leveraging external datasets, such as GTEx v8, DICE (Database of Immune Cell Expression)17, and the Human Plasma Proteome Atlas87,88.
Pleiotropic Associations with Disease
We explored pleiotropic associations between lead variants influencing antibody levels and several chronic diseases with known or hypothesized viral risk factors. Associations with selected cancers were obtained from a cancer pleiotropy meta-analysis of the UK Biobank and Genetic Epidemiology Research on Aging cohorts89. Summary statistics for the schizophrenia GWAS of 33,640 cases and 43,456 controls by Lam, Chen et al.90 were downloaded from the Psychiatric Genomics Consortium. Association p-values were obtained from the National Institute on Aging Genetics of Alzheimer’s Disease Data Storage Site for the GWAS by Jun et al.91, which included 17,536 cases and 53,711 controls. Associations with p<7.3×10−4 were considered statistically significant after correction for the number of variants and phenotypes tested.
HLA Regional Analysis
For phenotypes displaying a genome-wide significant signal in the HLA region, independent association signals were ascertained using two complementary approaches: clumping and conditional analysis. Clumping was performed on all variants with P<5×10−8 for each phenotype, as well as across phenotypes. Clumps were formed around sentinel variants with the lowest p-value and all other variants with LD r2>0.05 within a ±500kb window were assigned to that variant’s clump.
Next, we conducted conditional analyses using a forward stepwise strategy to identify statistically independent signals within each type of variant (SNP/indel or classical HLA allele). A total of 38,655 SNPs/indels on chromosome 6 (29,600,000 – 33,200,000 bp) were extracted to conduct regional analyses. Classical HLA alleles were imputed for UKB participants at 4-digit resolution using the HLA*IMP:02 algorithm applied to diverse population reference panels15. Imputed dosages were available for 362 classical alleles in 11 genes: HLA-A, HLA-B, and HLA-C (class I); HLA-DRB5, HLA-DRB4, HLA-DRB3, HLA-DRB1, HLA-DQA1, HLA-DQB1, HLA-DPA1, and HLA-DPB1 (class II). Analyses were restricted to 101 common alleles (frequency ≥ 0.01) in 413,810 European ancestry participants. Linear regression models were adjusted for the same get of covariates as the GWAS.
For each antigen response phenotype, we identified genetic variants (SNPs/indels or classical HLA alleles) with the lowest p-value and performed forward iterative conditional regression to identify other independent signals, until no associations with a conditional p-value (Pcond)<5×10−8 remained. Conditional analyses of classical HLA alleles were restricted to Bonferroni-significant associations corrected for 101 alleles (P<5×10−4) in the unconditional analysis. We also assessed the independence of associations across different types of genetic variants by including conditionally independent HLA alleles as covariates in the SNP-based analysis.
Genetic Associations With SARS-CoV-2 Status
GWAS of SARS-CoV-2 status was conducted using logistic regression with adjustment for age at assessment, sex, BMI, Townsend index, specimen origin (confirmed inpatient in acute care setting vs. unknown), genotyping array, and the top 10 PC’s. Models were also adjusted for smoking status to account for the possibility that more severe symptoms in smokers may increase their likelihood of SARS-CoV-2 testing. Based on recent evidence that SARS-CoV-2 uses the receptor encoded by the ACE2 gene for cell entry21, we systematically evaluated associations between significant ACE2 eQTLs (qFDR<0.05, P<4.0×10−4) identified in GTEx and SARS-CoV-2 test status. The relationship between genetic effects on ACE2 expression and SARS-CoV-2 status was investigated using a generalized estimating equations model, with log(OR) for testing positive as the outcome and normalized eQTL effect size (NES) as the predictor, clustered by tissue type. Lastly, for each classical HLA allele associated with at least one antigen response phenotype in common viruses (Bonferroni P<5×10−4), we tested its association with the likelihood of having a positive test result for SARS-Cov-2.
Transcriptome-Wide Association Analysis
Gene transcription levels were imputed and analyzed using the MetaXcan approach92, applied to GWAS summary statistics for quantitative antigen phenotypes. For imputation, we used biologically informed MASHR-M prediction models93 based on GTEx v8 with effect sizes computed using MASHR (Multivariate Adaptive Shrinkage in R)94 for variants fine-mapped with DAP-G (Deterministic Approximation of Posteriors)95,96. An advantage of this approach is that MASHR effect sizes are smoothed by taking advantage of the correlation in cis-eQTL effects across tissues. For each antigen, we performed a transcriptome-wide association study (TWAS) using gene expression levels in whole blood. Statistically significant associations for each gene were determined based on Bonferroni correction for the number of genes tested.
We also conducted sensitivity analyses based on gene expression profiles in tissues that represent known infection targets or related pathologies. Human herpes viruses and polyomaviruses are neurotropic and have been implicated in several neurological conditions97,98, therefore we considered gene expression in the frontal cortex. For EBV antigens additional models included EBV-transformed lymphocytes. Merkel cell polyomavirus (MCV) is known cause of Merkel cell carcinoma99, a rare but aggressive type of skin cancer, therefore we examined transcriptomic profiles in skin tissues for MCV only.
We summarized the pathways represented by genes associated with antibody response to viral antigens by conducting pathway enrichment analysis using curated Reactome gene sets and by examining protein interaction networks using the STRING database100Significantly associated TWAS genes were grouped by virus family (herpes viruses vs. polyomaviruses) and specificity of association (multiple antigens vs. single antigen). Protein interaction analyses were restricted to genes associated with more than one antigen. We considered unidirectional functional interactions with confidence scores ≥400 (medium).
DATA AVAILABILITY
The UK Biobank in an open access resource, available at https://www.ukbiobank.ac.uk/researchers/. This research was conducted with approved access to UK Biobank data under application number 14105 (PI: Witte).
URLs
PLINK 2.0: https://www.cog-genomics.org/plink/2.0/
R packages for pathway analysis: https://bioconductor.org/packages/release/bioc/html/ReactomePA.html and https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html
R package for protein network interaction analysis: http://xgr.r-forge.r-proiect.org
Data Availability
The UK Biobank in an open access resource, available at https://www.ukbiobank.ac.uk/researchers/. This research was conducted with approved access to UK Biobank data under application number 14105 (PI: Witte).
COMPETING INTERESTS
The authors declare no competing interests.
ACKNOWLEDGEMENTS
This research was supported by funding from the National Institutes of Health (US NCI R25T CA112355 and R01 CA201358; PI: Witte). Maike Morrison was funded by the University of California San Francisco’s Amgen Scholars Program.
References
- 1.↵
- 2.↵
- 3.↵
- 4.
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.
- 28.↵
- 29.↵
- 30.
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.↵
- 100.↵



