
Key Points
-
Type I interferon (IFN) responses are regulated by host, pathogen and environmental factors. These factors calibrate the host defences while limiting tissue damage and preventing autoimmunity.
-
Type I IFNs signal via the IFNα receptor (IFNAR) to activate receptor-associated Janus kinase 1 (JAK1) and tyrosine kinase 2 (TYK2) kinases and downstream signal transducer and activator of transcription (STAT) transcription factors; these transcription factors then induce the expression of IFN-stimulated genes (ISGs). Type I IFNs activate the IFN-stimulated gene factor 3 (ISGF3) complex, which is comprised of STAT1, STAT2 and IFN-regulatory factor 9 (IRF9); the ISGF3 complex binds to IFN-stimulated response elements (ISREs) to induce the expression of antiviral genes.
-
Type I IFN signalling is regulated in a quantitative and qualitative manner. The magnitude of signalling is increased by induction of STAT1 and IRF9 expression and amplification of JAK signalling by spleen tyrosine kinase (SYK) and protein tyrosine kinase 2 (PYK2). Conversely, the magnitude of signalling is decreased by suppressor of cytokine signalling (SOCS) and ubiquitin carboxy-terminal hydrolase 18 (USP18) proteins and by the downregulation and internalization of IFNAR. The qualitative nature of type I IFN responses is determined by the balance between the activation of various STATs and ISGF3.
-
Type I IFN-induced transcription is regulated by the post-translational modification of STATs, chromatin remodelling, the epigenetic landscape and cooperation with other transcription factors, co-activators and co-repressors.
-
ISGs encode proteins that regulate the translation of IFNAR and JAK–STAT signalling components and of ISGs themselves. Type I IFNs also induce the expression of microRNAs that regulate the IFN response.
-
Chronic type I IFN responses can promote autoimmunity by increasing antigen presentation, lymphocyte-mediated adaptive immune responses and chemokine expression. In chronic infections, type I IFNs can induce immunosuppression in part by increasing expression of interleukin-10 and programmed cell death 1 ligand 1 (PDL1).
Abstract
Type I interferons (IFNs) activate intracellular antimicrobial programmes and influence the development of innate and adaptive immune responses. Canonical type I IFN signalling activates the Janus kinase (JAK)–signal transducer and activator of transcription (STAT) pathway, leading to transcription of IFN-stimulated genes (ISGs). Host, pathogen and environmental factors regulate the responses of cells to this signalling pathway and thus calibrate host defences while limiting tissue damage and preventing autoimmunity. Here, we summarize the signalling and epigenetic mechanisms that regulate type I IFN-induced STAT activation and ISG transcription and translation. These regulatory mechanisms determine the biological outcomes of type I IFN responses and whether pathogens are cleared effectively or chronic infection or autoimmune disease ensues.
Similar content being viewed by others

Main
Type I interferons (IFNs) are polypeptides that are secreted by infected cells and have three major functions. First, they induce cell-intrinsic antimicrobial states in infected and neighbouring cells that limit the spread of infectious agents, particularly viral pathogens. Second, they modulate innate immune responses in a balanced manner that promotes antigen presentation and natural killer cell functions while restraining pro-inflammatory pathways and cytokine production. Third, they activate the adaptive immune system, thus promoting the development of high-affinity antigen-specific T and B cell responses and immunological memory (Fig. 1). Type I IFNs are protective in acute viral infections but can have either protective or deleterious roles in bacterial infections and autoimmune diseases1.

On pathogen detection, infected cells produce type I interferons (IFNs). Innate immune cells, such as macrophages and dendritic cells (DCs), produce type I IFNs after sensing pathogen components using various pattern-recognition receptors (PRRs), which are found on the plasma membrane, in endosomes and throughout the cytosol. In particular, plasmacytoid DCs (pDCs) produce large quantities of IFNα. Non-immune cells, such as fibroblasts and epithelial cells, predominantly produce IFNβ. In infected and neighbouring cells, type I IFNs induce the expression of IFN-stimulated genes (ISGs), the products of which initiate an intracellular antimicrobial programme that limits the spread of infectious agents. Innate immune cells also respond to type I IFNs by enhancing antigen presentation and the production of immune response mediators, such as cytokines and chemokines. Adaptive immunity is also affected by type I IFNs: for example, type I IFNs can augment antibody production by B cells and amplify the effector function of T cells. PAMP, pathogen-associated molecular pattern.
This Review focuses on the most well-defined type I IFNs, namely IFNα and IFNβ (the remaining type I IFNs have been reviewed elsewhere)2,3. Most cell types produce IFNβ, whereas haematopoietic cells, particularly plasmacytoid dendritic cells, are the predominant producers of IFNα. IFNβ is encoded by a single IFNB gene, whereas 14 distinct genes encode various IFNα isoforms. Type I IFN production is induced after the sensing of microbial products by pattern-recognition receptors (PRRs)4,5,6 and by cytokines. Recent developments that extend our understanding of type I IFN production are summarized in Box 1.
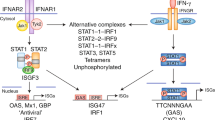
IFNα and IFNβ bind a heterodimeric transmembrane receptor termed the IFNα receptor (IFNAR), which is composed of IFNAR1 and IFNAR2 subunits. In the canonical type I IFN-induced signalling pathway described over 25 years ago, IFNAR engagement was shown to activate the receptor-associated protein tyrosine kinases Janus kinase 1 (JAK1) and tyrosine kinase 2 (TYK2), which phosphorylate the latent cytoplasmic transcription factors signal transducer and activator of transcription 1 (STAT1) and STAT2 (Refs 7, 8) (Fig. 2). Tyrosine-phosphorylated STAT1 and STAT2 dimerize and translocate to the nucleus, where they assemble with IFN-regulatory factor 9 (IRF9) to form a trimolecular complex called IFN-stimulated gene factor 3 (ISGF3). ISGF3 binds to its cognate DNA sequences, which are known as IFN-stimulated response elements (ISREs; consensus sequence TTTCNNTTTC), thereby directly activating the transcription of ISGs. By contrast, most other cytokines activate STAT homodimers that bind to a distinct gamma-activated sequence (GAS; consensus sequence TTCNNNGAA). Thus, canonical type I IFN signalling induces a distinct subset of several hundred ISRE-driven ISGs, many of which establish a cellular antiviral state8,9,10,11. ISG-encoded proteins restrain pathogens by several mechanisms, including the inhibition of viral transcription, translation and replication, the degradation of viral nucleic acids and the alteration of cellular lipid metabolism9,12.

On engagement, the interferon-α receptor (IFNAR, which is composed of the IFNAR1 and IFNAR2 subunits) activates Janus kinase 1 (JAK1) and tyrosine kinase 2 (TYK2). Phosphorylation of the receptor by these kinases results in the recruitment of signal transducer and activator of transcription (STAT) proteins, phosphorylation, dimerization and nuclear translocation. The three predominant STAT complexes that are formed in response to type I interferon (IFN) control distinct gene-expression programmes. The interferon-stimulated gene factor 3 (ISGF3) complex (which is composed of STAT1, STAT2 and IFN-regulatory factor 9 (IRF9)) binds to IFN-stimulated response element (ISRE) sequences to activate classical antiviral genes, whereas STAT1 homodimers bind to gamma-activated sequences (GASs) to induce pro-inflammatory genes. STAT3 homodimers indirectly suppress pro-inflammatory gene expression, probably by the induction of as-yet-unknown transcriptional repressors. Type I IFN-activated STAT3 is bound by the co-repressor complex SIN3 transcription regulator homologue A (SIN3A), which suppresses induction of direct STAT3 target genes by promoting de-acetylation of STAT3 and histones. Type I IFNs also activate STAT4 and can activate STAT5 and STAT6 in a context-dependent manner (not depicted). CXCL9, CXC-chemokine ligand 9; MX1, IFN-induced GTP-binding protein Mx1; OAS, 2′-5′-oligoadenylate synthase; P, phosphate.
Cellular responses to IFNAR ligation are cell type- and context-dependent and vary during the course of an immune response13. This variation results from cell lineage and developmental differences that alter how cells interpret IFNAR-induced signals, and from the modulation of IFNAR signalling and ISG expression by concurrent signals induced by pathogens, PRRs and immune and environmental factors. These heterologous signals regulate cell responses to IFNAR ligation at several levels. First, they regulate the expression and post-translational modification of IFNAR and JAK–STAT signalling components, thus modulating the magnitude and pattern of IFNAR signalling. Second, they induce transcription factors that cooperate with STATs and modulate chromatin states at target gene loci, thus specifying downstream ISG expression. Third, they modify factors that mediate translation, thus regulating the pattern of protein expression. Type I IFN responses are fine-tuned by opposing augmenting and suppressive signals induced by host factors; these signals rapidly mobilize an effective antimicrobial response against the invading pathogen while restraining the magnitude of the response to avoid attendant toxicity. Pathogens can use some of the same suppressive mechanisms to evade the antimicrobial actions of type I IFNs. This Review focuses on how host, pathogen and environmental factors cross-regulate IFNAR-induced signalling, transcription and translation to determine the biological outcome of type I IFN signalling.
Regulation of type I IFN signalling
Basal type I IFN signalling. The canonical type I IFN signalling pathway components IFNAR, JAK1, TYK2, STAT1, STAT2 and IRF9 are widely expressed, and thus most cell types are competent to mount type I IFN-dependent responses. Immune cells can respond rapidly to low levels of type I IFNs, a capacity that is maintained under homeostatic conditions by an autocrine loop in which small amounts of IFNβ maintain high basal expression levels of STAT1 and IRF9, which are themselves ISGs14. Basal IFNβ expression and attendant tonic IFNAR signalling equips immune cells to rapidly mobilize effective antimicrobial programmes. Recent work suggests that basal IFNβ production under homeostatic conditions is driven in part by the commensal microbial flora, which thus can systemically calibrate type I IFN responses15,16,17. In addition, IFNAR signalling is augmented by tonic signals generated by immunoreceptor tyrosine-based activation motif (ITAM)-associated immunoreceptors that bind constitutively expressed endogenous ligands18. Tonic ITAM signalling activates protein tyrosine kinase 2 (PYK2; also known as PTK2) to amplify type I IFN-induced JAK activation. At the same time, the magnitude of basal IFNAR signalling is restrained by opposing mechanisms that limit expression of IFNAR–JAK–STAT signalling components. These suppressive mechanisms include the pausing of RNA polymerase II (Pol II) at genes that encode IFN pathway components19 and the induction of microRNAs (miRNAs) that destabilize or suppress translation of the corresponding transcripts20,21,22,23. During the course of an immune response, cell responses to newly produced type I IFNs are modified by feedback mechanisms that are activated by type I IFNs and by heterologous signals that are generated by microbial factors and host cytokines.
Augmentation of type I IFN signalling. Mechanisms that amplify type I IFN signalling include the induction of STAT1 and IRF9 expression, the augmentation of STAT tyrosine phosphorylation by spleen tyrosine kinase (SYK) and PYK2 and the enhancement of STAT1 transcriptional activity by serine phosphorylation (Fig. 3). STAT1 and IRF9 are ISGs, and their expression is induced by type I IFNs, IFNγ and other cytokines that activate STATs, such as interleukin-6 (IL-6)24,25,26,27. Increased levels of STAT1 lead to its interaction with and activation by IFNAR, whereas increased levels of IRF9 promote the binding of ISGF3 to target genes. Notably, low picomolar concentrations of IFNs are sufficient to increase STAT1 expression, thereby priming macrophages for enhanced responsiveness to type I IFNs during the early phases of infection, when the amounts of this cytokine are limiting. This priming mechanism is also operative during tumour necrosis factor (TNF)-induced activation of macrophages, in which TNF induces picomolar concentrations of IFNβ that increase STAT1 expression, resulting in high induction of ISGs28,29. High expression of STATs and IRF9 can sustain expression of a subset of ISGs over prolonged time periods, even in the absence of on-going cytokine-mediated signalling and tyrosine phosphorylation of JAKs and STATs30. In addition to increasing STAT expression, the priming of macrophages with low concentrations of IFNγ functionally couples the SYK kinase with IFNAR to amplify signalling and STAT1 activation; this enhancement is dependent on signalling by ITAM-associated immunoreceptors26. Phosphorylation of a conserved carboxy-terminal serine in STAT1 that leads to enhanced transcriptional activity is mediated by various members of the protein kinase C (PKC) and mitogen-activated protein kinase (MAPK) families8. Thus, STAT1 transcriptional activity and downstream ISG induction are modulated by a large number of host factors that activate PKCs and MAPKs, including inflammatory cytokines and chemokines. Overall, IFNAR signalling is augmented via distinct mechanisms by major families of heterologous receptors that are engaged by various ligands during immune responses. This enhances the host-mediated mobilization of type I IFN responses during infections and promotes the antimicrobial and pro-inflammatory functions of type I IFNs.

Various receptor pathways cross-regulate the type I interferon (IFN) response; this alters the expression levels and activation states of IFN signalling components. Various mitogen-activated protein kinase (MAPK)-activating pathways, such as those induced by pattern- recognition receptors (PRRs), enhance signal transducer and activator of transcription 1 (STAT1) transcriptional activity through phosphorylation of a conserved carboxy-terminal serine. Low-level basal signalling by immunoreceptor tyrosine-based activation motif (ITAM)-containing receptors also augments Janus kinase 1 (JAK1) activity through activation of spleen tyrosine kinase (SYK) and protein tyrosine kinase 2 (PYK2). Conversely, strong activation of ITAM-containing receptors by high-avidity crosslinking suppresses IFNα receptor (IFNAR) signalling via protein kinase C (PKC)-mediated recruitment of SH2 domain-containing protein-tyrosine phosphatase 2 (SHP2), which dephosphorylates signalling intermediates. Cytokine signalling pathways, including the interleukin-1 (IL-1) pathway, inhibit type I IFN responses by directly promoting IFN receptor turnover via p38 kinase and casein kinase II (CK2). Various cytokines that signal through JAK–STAT pathways, including type I IFNs, regulate the expression levels of both positive and negative regulators of the IFN response pathways. JAK–STAT-induced positive regulators include STAT1 and IFN-regulatory factor 9 (IRF9), whereas negative regulators include the suppressor of cytokine signalling (SOCS) protein family and ubiquitin carboxy-terminal hydrolase 18 (USP18). Inhibitor of NF-κB kinase-ε (IKKε)-mediated STAT1 phosphorylation inhibits STAT1 homodimerization, thereby promoting activation of the IFN-stimulated gene factor 3 (ISGF3) complex rather than STAT1 homodimers in response to type I IFN. IKKε expression is induced by various pathways, including tumour necrosis factor (TNF)- and PRR-induced pathways. MicroRNAs induced by cytokine and PRR pathways downregulate the expression of IFN response signalling proteins: for example, miR-146a suppresses STAT1 expression, and miR-155 causes a collective reduction in various IFN signalling proteins. AGO2, Argonaute 2; RISC, RNA-induced silencing complex.
Suppression of type I IFN signalling. Mechanisms that suppress type I IFN-mediated responses include downregulation of cell surface IFNAR expression, induction of negative regulators (such as suppressor of cytokine signalling (SOCS) proteins and ubiquitin carboxy-terminal hydrolase 18 (USP18), and the induction of miRNAs (Fig. 3). Internalization of IFNAR is induced by various heterologous pathways to limit cellular IFN responsiveness31,32. These include signalling pathways activated by pro-inflammatory cytokines such as IL-1, Toll-like receptors (TLRs), ITAM-associated receptors and oxidative or metabolic stress31,33,34,35. One inhibitory mechanism is p38 kinase-mediated priming phosphorylation of IFNAR that facilitates subsequent casein kinase II (CK2)-mediated phosphorylation of a degron sequence in the IFNAR cytoplasmic domain, thereby increasing receptor internalization, ubiquitylation and degradation31. Viruses and tumour cells also induce IFNAR degradation to evade type I IFN-mediated antiviral responses and antitumour effects, respectively31,36. In addition to inducing IFNAR internalization, TLR stimulation and high-avidity crosslinking of ITAM-associated receptors can also suppress IFNAR signalling by recruiting PKCβ and PKCδ to IFNAR; the activation of SH2 domain-containing protein tyrosine phosphatase 2 (SHP2; also known as PTPN11), which is associated with this recruitment, can dephosphorylate signalling intermediates34,37,38.
Type I IFNs induce SOCS1, SOCS3 and USP18 as part of a negative feedback loop to limit the extent and duration of type I IFN responses39,40. SOCS proteins compete with STATs for binding to IFNAR and suppress JAK activity, whereas USP18 displaces JAK1 from IFNAR2. These inhibitory proteins are also induced by various cytokines that activate JAK–STAT signalling, by ITAM-associated receptors (including dectin 1 (also known as CLEC7A), which senses fungal pathogens) and by pro-inflammatory cytokines and PRRs that activate MAPK pathways. Thus, pathogens can use these negative regulators to escape from type I IFN-mediated antimicrobial actions. Type I IFN responses are further regulated by miRNAs41. STAT1 expression in T helper 1 (TH1) cells is tightly regulated by miR-146a21,23. miR-155 is highly induced during cell activation and by pro-inflammatory and PRR signalling22,42, and a recent report shows that miR-155 broadly suppresses expression of IFNAR–JAK–STAT pathway components in CD8+ T cells. This suppression enhances CD8+ T cell responses to viral and bacterial pathogens by downregulating the negative effects of type I IFNs on T cell proliferation22.
Qualitative regulation of type I IFN signalling. In addition to its magnitude, the qualitative nature of IFNAR signalling is regulated, thus enabling distinct biological outcomes downstream of a common proximal signal. One mechanism that explains how IFNAR signalling can activate different patterns of downstream gene expression is the differential activation of STATs that have distinct target genes and biological functions (Fig. 2). In addition to the activation of ISGF3 that was described originally, IFNAR activates STAT1 and STAT3 homodimers and heterodimers in most cell types and can activate STAT4, STAT5 and STAT6 in certain cell types13. The balance between activation of different STATs by IFNAR is partially determined by relative STAT expression levels. Thus, preferential induction of STAT1 expression by IFNs and cytokines, as occurs in the priming systems described above, is associated with increased type I IFN-induced activation of STAT1 homodimers and expression of GAS-containing pro-inflammatory genes (similar to those activated by IFNγ)25,26,27. In contrast to STAT1, STAT3 activates genes that suppress inflammatory responses43, suggesting that type I IFNs can activate a parallel STAT3-mediated signalling pathway that balances and restrains the pro-inflammatory pathways induced by type I IFNs. This notion is supported by a study in which the relative expression of STAT1 and STAT3 was modulated using complementary forced expression and RNA interference approaches. This study found that relative expression of STATs is an important determinant of the pattern of IFNAR-induced STAT activation and showed that STAT3 restrains STAT1-mediated inflammatory signalling downstream of IFNAR44. Similarly, experiments in which STAT3 was deleted in myeloid cells showed that STAT3 restrains IFNAR-induced antiviral responses45. The idea that shifts in the balance of activation of distinct STAT complexes downstream of IFNAR mediates its context-dependent function by altering the balance between antiviral, pro-inflammatory, suppressive and anti-proliferative functions is supported by studies showing a switch in IFNAR signalling from activation of STAT1 to activation of STAT4 in CD8+ T cells during lymphocytic choriomeningitis virus (LCMV) infection46,47. This switch in STAT activation enables optimal antigen-specific CD8+ T cell expansion and promotes immunity to LCMV.
Mechanisms other than relative expression levels regulate the pattern of IFNAR-induced STAT activation. For example, the balance between ISGF3 and STAT1 homodimer activation is also regulated by inhibitor of NF-κB kinase-ε (IKKε)48,49. IKKε-mediated phosphorylation of STAT1 suppresses STAT1 homodimer formation, thereby facilitating ISGF3 formation and augmenting antiviral responses. It will be interesting to determine whether activation of IKKε by TLRs or its induction by TNF affects the patterns of ISG expression that are induced by these factors28. In addition, changes in the relative expression of different STATs are insufficient to explain how switching from STAT1 to STAT4 activation by IFNAR occurs in CD8+ T cells during LCMV infection46. Increased STAT1 expression is also insufficient to explain the strong shift to STAT1 homodimer activation that occurs in IFN-primed macrophages; in this case, SYK contributes to preferential tyrosine phosphorylation of STAT1 (Ref. 26).
Concurrent stimuli can also potentially couple IFNAR to new signalling pathways. For example, in the context of infection with Salmonella enterica subsp. enterica serovar Typhimurium, IFNAR directly interacts with and activates receptor-interacting serine/threonine-protein kinase (RIP) kinases and thus can induce RIP-mediated cell death50. This S. Typhimurium-induced change in IFNAR signalling seems to favour the persistence of the pathogen because it allows this intracellular bacterium to evade innate immune responses through direct elimination of innate immune cells. It is also possible that signalling by heterologous receptors affects a newly described signalling pathway in which IFNβ binds directly to IFNAR1, without a requirement for IFNAR2, and activates a novel JAK–STAT-independent gene response51.
Regulation of type I IFN-induced transcription
In the classical model of JAK–STAT signalling, the predominant determinant of gene activation is the nuclear translocation of STATs and their direct binding to cognate GAS elements (Fig. 2); in the case of type I IFNs, this includes the formation of ISGF3 and its binding to ISREs7. It has become clear that this model is incomplete and that STAT- and ISGF3-induced gene transcription is regulated at several levels: first, by post-translational modification of STATs in the nucleus; second, through the cooperation of STATs with other transcription factors; third, by epigenetic factors and chromatin states at target gene loci that modulate the access of STATs to their target genes; fourth, through the interaction of STATs with co-activators, co- repressors, chromatin-modifying complexes and transcription elongation factors; and fifth, by the binding of STATs to distal regulatory elements, such as enhancers. These regulatory mechanisms and their modulation by heterologous signalling pathways are discussed in the following section.
Post-translational modification. As described above, tyrosine and serine phosphorylation of STATs occurs in the cytoplasm, but serine phosphorylation of STATs also occurs in the nucleus, presumably at target gene loci52. This phosphorylation event is mediated in part by cyclin-dependent kinase 8 (CDK8; which is a component of the Mediator co-activator complex) after STAT1 binds DNA52,53. CDK8 modulates IFNγ-induced gene expression and augments antiviral responses, but its functional role in IFNAR signalling is not yet known. STATs are also acetylated, methylated and sumoylated8,54,55,56; it will be important to elucidate how these modifications are regulated and how they modify IFNAR-induced gene activation.
Cooperation with other transcription factors. The most established interaction of STATs with other transcription factors has been shown for members of the IRF family57. The DNA sequences recognized by most IRFs overlap with the ISRE, and thus IRFs can bind to and activate many of the same target genes that ISGF3 does. In addition, expression of IRF1, IRF7, IRF8 and IRF9 is induced by IFNs through STAT-dependent mechanisms, and many ISGs contain both GAS and ISRE or IRF elements. This has led to a paradigm of IFN-induced gene expression in which STATs and ISGF3 directly activate certain ISGs, including IRF1, IRF7, IRF8 and IRF9, and these IRFs activate a second group of ISGs, the induction of which is dependent on de novo protein synthesis13. Induction of IRF7 expression leads to a feedforward loop in which subsequent post-translational activation of IRF7 by PRR signalling drives downstream expression of IFNA and various target genes58,59. Importantly, STATs, ISGF3 and IRFs cooperate in the induction of many ISGs by coordinated binding to cognate sequences in gene promoters. Recent work has shown genome-wide co-occupancy of promoters and enhancers by STAT1 and IRF1 after stimulation with IFNγ or lipopolysaccharide (LPS, which induces autocrine IFNβ production), suggesting that the extensive cooperation between these factors extends beyond the induction of classical ISGs60. STATs can associate with IRFs to form protein complexes that are distinct from ISGF3, as STAT1 associates with IRF1 and can therefore bind indirectly to gene regulatory elements that do not contain GAS sites60,61.
STATs also cooperate with transcription factors that are activated by heterologous signalling pathways: for example, STATs interact with NF-κB, which is activated by multiple PRRs and inflammatory cytokines13. This enables increased or synergistic induction of target genes and is mediated in part by coordinate binding of STATs and NF-κB to GAS and NF-κB sites in target gene promoters. Recently, Decker and colleagues described a new mechanism by which PRR and type I IFN-mediated signals cooperate to induce expression of nitric oxide synthase 2 (Nos2) during Listeria monocytogenes infection. PRR-mediated activation of NF-κB primes the Nos2 promoter by recruiting the basal transcription factor TFIIH and the Pol II activating kinase CDK7 (Ref. 62). Subsequently, autocrine IFNβ activates ISGF3, which recruits Pol II to the Nos2 promoter, where Pol II is phosphorylated and activated by NF-κB-recruited CDK7.
Promyelocytic leukaemia zinc finger protein (PLZF; also known as ZBTB16) has emerged as a transcription factor that is required for the expression of a subset of ISGs, and it is particularly important for natural killer cell function in the context of viral infections. PLZF transcriptional activation of ISGs depends in part on recruitment of histone deacetylase 1 (HDAC1) and serine phosphorylation by the extracellular signal- regulated kinase (ERK) family of MAPKs63. ERK-dependent phosphorylation of PLZF is induced in some cell types (such as HeLa cells) by IFNs and might also mediate cross-regulation of ISG induction in immune cells by heterologous pathways that activate MAPKs.
Type I IFN responses can also be inhibited by transcriptional repressors. A recent report identified DNA regulatory elements for the transcription factor forkhead box protein O3 (FOXO3) in several ISG promoters, including IRF7 (Ref. 64). On the basis of experiments with genetically altered mice and viral infections, the authors proposed that FOXO3 functions to repress basal type I IFN responses and also shuts down the type I IFN response during the resolution phase of an infection. Relatively little is known about the repression of ISG expression, and the characterization of additional repressors represents an important area for future study.
Even in populations of nearly identical cells, not all cells that are infected by a virus or exposed to microbial products produce type I IFNs and express ISGs65,66,67. The stochastic nature of ISG expression seems to be related to bimodal induction of STAT2 and IRF7, which can drive high amounts of ISG transcription in a particular subset of cells, whereas other cells express much lower amounts of ISG transcripts. The biological purpose of stochasticity is unclear, but some evidence suggests that it generates cells that are 'mass IFN producers'. These mass IFN producers help to combat the infection but might also be more susceptible to cell death, while 'low IFN producers' are preserved in order to maintain the viability of tissues.
Chromatin remodelling. Gene expression is restrained by nucleosome barriers, in which histone octamers bind the DNA and prohibit transcription factor binding and Pol II extension68,69. Before gene expression can be induced, this barrier must be overcome through the post-translational modification of histones (for example, by acetylation that 'loosens' the interaction of octamers with DNA) and through the repositioning or removal of nucleosomes by ATP-dependent remodelling complexes (Fig. 4). The induction of ISGs by type I IFNs requires chromatin remodelling, which occurs via STAT1-, STAT2- and IRF-mediated recruitment of nucleosome-remodelling enzymes and histone acetyltransferases (HATs). Early work in cell lines showed that type I IFN stimulation results in recruitment to ISGs of BAF, an ATP-dependent nucleosome remodelling complex (which corresponds to yeast SWI/SNF)70,71,72,73,74. Recruitment of BAF is mediated at least in part by the association of STAT2 with the BRG1 subunit of BAF, which results in increased accessibility at ISG loci and correlates with increased transcription of ISGs70,71,72,73,74. BAF is also important for the induction of a subset of ISGs and other secondary response genes after TLR-mediated activation of myeloid cells75. Transcriptome analysis suggests that the low abundance of CpG islands in ISG promoters predisposes to high nucleosome occupancy and confers a requirement for nucleosome remodelling76,77.

In unstimulated cells, interferon-stimulated gene (ISG) transcription is suppressed by repressive transcription factors such as forkhead box protein O3 (FOXO3), high nucleosome occupancy due to low GC nucleotide content and repressive complexes bound to methylated histones. On type I interferon (IFN) stimulation, the IFN-stimulated gene factor 3 (ISGF3) complex (which contains signal transducer and activator of transcription 1 (STAT1), STAT2 and IFN-regulatory factor 9 (IRF9)) binds to ISG promoters and enhancers, recruiting various chromatin remodelling and transcriptional activator complexes. These complexes include the CREB-binding protein (CBP) histone acetyltransferase, the BRG1 chromatin remodelling factor, the multi-subunit Mediator co-activator complex, the Pol II-associated factor 1 homologue (PAF1) elongation factor and bromodomain-containing proteins (BRDs) such as BRD4 that bind to acetylated histones. BRDs recruit positive transcription elongation factor b (pTEFb), which augments transcription by phosphorylating RNA polymerase II (Pol II). CDK8, cyclin-dependent kinase 8; EHMT1, euchromatic histone-lysine N-methyltransferase 1; H4ac, histone H4 acetylation; H3K9me2; histone H3 lysine 9 dimethylation.
Histone lysine acetylation, particularly on histones H3 and H4 in nucleosomes in the vicinity of regulatory regions, is associated with transcriptional activity78. On induction, ISG promoters associate with HATs and exhibit increased levels of histone acetylation; this acetylation is mediated in part by the recruitment of the HATs p300, CREB-binding protein (CBP) and GCN5 (also known as KAT2A) through interaction with the transcription activation domains of STAT1 and STAT2 (Ref. 8). Recent reports have linked the STAT1 binding that occurs after IFNγ-mediated stimulation with histone acetylation at promoters and enhancers genome wide60,79,80, and it is likely that STATs mediate extensive histone acetylation after type I IFN stimulation as well. One mechanism by which acetylated histones increase transcription is through the recruitment of bromodomain (BRD)-containing proteins such as BRD4, which in turn recruit the positive transcription elongation factor b (pTEFb) complex to promote transcription elongation81 (Fig. 4). Consistently, transcription of ISGs is sensitive to inhibitors, such as I-BET and JQ1, that block the interaction of bromodomains with acetylated histones82,83, although direct interaction of BRDs with pTEFb at ISGs has not been demonstrated, and BRDs might promote ISG transcription by alternative mechanisms. Paradoxically, ISG induction can also be blocked by inhibitors of HDACs, but this blockade is indirect and is likely to be caused by HDAC inhibition at non-ISG loci84. Type I IFNs also induce H2B ubiquitylation, a positive mark that promotes type I IFN-driven ISG transcription and is mediated by the E3 ubiquitin ligase BRE1 (Ref. 85). Adenoviruses evade the type I IFN response by disrupting the BRE1 complex and thus suppressing ISG expression85.
Similarly to other transcription factors, ISGF3 interacts with co-activator or co-repressor proteins that mediate interactions with the general transcriptional machinery and also facilitate the recruitment of enzymes that modify chromatin: for example, HATs, HDACs and histone methyltransferases. STAT1 interacts with the CDK8 subunit of the Mediator co-activator complex, whereas STAT2 within ISGF3 binds to subunit 14 of Mediator (MED14; also known as DRIP150) and to RVB proteins53,86,87. The interaction with RVB proteins is required for type I IFN-induced histone acetylation and Pol II recruitment to various ISG promoters87. IRF9 within ISGF3 complexes interacts with glucocorticoid receptor-interacting protein 1 (GRIP1; also known as NCOA2), which is best known as a co-regulator of the glucocorticoid receptor, to promote ISG expression88. This interaction enables glucocorticoids to suppress ISG expression by binding GRIP1 and displacing it from ISGF3. IFNα-activated STAT3 binds the co-repressor complex SIN3 transcription regulator homologue A (SIN3A), which contains HDAC1 and HDAC2 and suppresses STAT3-mediated gene induction89 (Fig. 2). This explains the long-standing puzzle of why STAT3 target genes are not activated by type I IFNs in certain cell types, despite robust STAT3 activation. SIN3A knockdown, which allows type I IFN-activated STAT3 to function, results in diminished expression of ISGs, supporting the notion discussed above that STAT3 can counterbalance type I IFN-induced STAT1 and ISGF3 function89. Protein inhibitor of activated STAT1 (PIAS1) is a small ubiquitin-related modifier (SUMO) ligase that can suppress STAT1 function and ISG expression by suppressing the binding of STAT1 to DNA and by acting as a co-repressor to recruit 'writers' of negative histone marks90,91,92.
Transcription elongation. Transcriptional elongation of ISGs is also regulated. After type I IFN-mediated stimulation, negative elongation factor (NELF) and DRB sensitivity-inducing factor (DSIF) proteins that promote the pausing of Pol II are recruited to ISGs to restrain transcriptional output83. Transcriptional elongation at ISG loci is promoted by the Pol II-associated factor 1 homologue (PAF1) complex, which is disrupted by the influenza protein NS1 to facilitate evasion of the antiviral type I IFN programme by this pathogen93.
Epigenetic landscape. STATs and ISGF3 can remodel chromatin to activate regulatory elements, but what determines where STATs bind in the first place? The current paradigm is that during cell differentiation, lineage-determining transcription factors create a cell type-specific regulatory landscape by allowing access to certain regulatory elements (promoters and enhancers); in turn, the regulatory landscape of accessible chromatin determines the binding pattern of signal-activated factors, such as STATs94. The enhancer repertoire is cell type specific and thus can control the initial pattern of ISG induction, although this regulatory mechanism has not yet been directly tested for type I IFN responses. Recently it has become clear that STAT1 primes and/or activates enhancers genome wide during the differentiation or polarization of T cells and macrophages60,79, and can contribute to the creation of new enhancers in differentiated cells80. Thus, STAT1 can alter the epigenetic landscape, suggesting that IFNs have the potential to alter cell responses to subsequent stimuli. Type I IFN-induced ISG expression is also controlled quantitatively in a cell lineage-specific manner by the negative histone mark histone H3 lysine 9 dimethylation (H3K9me2), which attenuates gene expression. Deposition of H3K9me2 at ISGs by the G9A methyltransferase results in a weaker type I IFN response in fibroblasts than in haematopoietic cells, where H3K9me2 marks at ISGs are at low levels or absent95. Furthermore, the G9A homologue euchromatic histone-lysine N-methyltransferase 1 (EHMT1; also known as GLP) catalyses H3K9 methylation at ISGs to suppress transcription, and knockdown of EHMT1 augments antiviral immunity96.
Translational regulation of type I IFN responses
A well-established antiviral mechanism of type I IFNs is the global suppression of translation, which serves to inhibit the production of viral proteins and thus viral replication. Type I IFNs suppress translation in part by inducing ISGs that directly suppress translation10, such as protein kinase RNA-activated (PKR; also known as eIF2AK2), which phosphorylates and inactivates the key translation factor eukaryotic translation initiation factor 2A (eIF2A). Type I IFN-induced suppression of translation and its role in antiviral responses has been reviewed97; here we focus on how translational regulation affects the type I IFN response by modulating the expression of STAT proteins and ISGs. A key question is how the translation of effectors of host defence, such as ISGs, is maintained and regulated in the setting of globally suppressed translation.
Although type I IFNs broadly suppress translation, in certain cell types they activate phosphoinositide 3-kinase (PI3K)–AKT–mammalian target of rapamycin (mTOR) signalling64,98,99,100,101, a pathway that strongly promotes translation by inactivating the translational suppressor eIF4E-binding protein 1 and activating p70S6 kinase, which in turn activates eIF4 translation initiation factors. Although mTOR signalling typically selectively promotes the translation of transcripts containing conserved terminal oligopyrimidine (TOP) and/or pyrimidine-rich translational element (PRTE) sequences in their 5′ untranslated regions (UTRs)102,103, IFNAR–mTOR signalling also promotes translation of select ISGs via activation of p70 S6 kinase and eIF4 translation initiation factors99,100. This pathway might contribute to increased IRF7 translation, with attendant downstream induction IFNα production104. Type I IFNs also increase the frequency of cycles of translational activity and arrest that occur in cells that are infected with RNA viruses that activate PKR, and this activity seems to facilitate the translation of host proteins that are important for cell survival105.
IFNβ induces the expression of various miRNAs that can contribute to an antiviral state by targeting viral and possibly host transcripts, resulting in decreased translation and increased degradation (reviewed in Ref. 23). Several cellular miRNAs that target STAT1, STAT2 and IFNβ transcripts are induced by type I IFNs, including miR-146a, which is also induced by PRR and inflammatory cytokine signalling21,23. Thus, it seems that type I IFN-induced miRNAs participate in feedback inhibition and can also be induced by other factors to cross-regulate type I IFN responses. The regulation of type I IFN responses by miRNAs merits additional investigation, and a substantial advance has been the identification of host transcripts that are directly targeted by type I IFN-induced miRNAs. This was achieved by using the HITS-CLIP technique to identify transcripts that are associated with RNA-induced silencing complexes (P. Herztog, unpublished observations).
ISG-encoded proteins can inhibit translation broadly or target specific transcripts10. Widespread suppression of general translational machinery has been documented for IFN-induced protein with tetratricopeptide repeats 1 (IFIT1) and IFIT2, which bind and inhibit eIF3-mediated formation of the ribosomal pre-initiation complex106,107, whereas PKR phosphorylates and inhibits the eIF2 complex, which is important for ribosomal function. The general translational repressor 4E-BP, which is regulated by type I IFN–mTOR signalling, can suppress IRF7 translation104, which might serve as a preventive measure to limit IRF7 expression in the absence of positive regulatory signals. In addition, IRF7 translation is suppressed by the fellow ISG 2′-5′-oligoadenylate synthase-like protein 1 (OASL1) in an inducible and highly specific manner. Although it does not affect translation globally, OASL1 specifically suppresses IRF7 translation by binding to a double stem–loop structure in the IRF7 5′UTR108. Thus, the translation of ISGs during a type I IFN response can be regulated by unique sequences that are present in their mRNAs. OASL1 expression increases after the stimulation of type I IFNs, so this represents a negative feedback pathway that limits the duration or intensity of the type I IFN response.
Regulation of chronic IFN responses
The antiviral and immune stimulatory properties of type I IFNs during acute viral infections are well established, and their dual protective and deleterious roles in acute bacterial infections have also been reviewed1. Less is known about the role of the chronic increase of type I IFNs and the associated type I IFN response (which is observed as an 'IFN signature' of ISG expression) in various autoimmune diseases and chronic viral and mycobacterial infections. A chronic type I IFN response is associated with the autoimmune diseases systemic lupus erythematosus (SLE), Sjogren's syndrome, systemic sclerosis, myositis and rheumatoid arthritis109. Disease-associated allelic variants in IFN pathway genes and in ISRE sequences that are present in enhancers and promoters genome wide also link the IFN pathway with these diseases110,111. A type I IFN signature is detected in patients who are chronically infected with hepatitis C virus (HCV) and HIV112, and more recently it was described and, surprisingly, linked with increased disease activity in the chronic mycobacterial diseases tuberculosis and lepromatous leprosy113,114,115,116. In this section, we review the potential role of type I IFN responses in these settings and discuss how the complex milieu of these chronic diseases can regulate type I IFN responses to affect disease pathogenesis. Type I IFNs also have an important role in anticancer resistance, which will not be reviewed because the regulation of IFN responses in this context has not been well established.
Functions of type I IFNs in autoimmune diseases. Generally, type I IFNs are thought to have a pathogenic role in autoimmune diseases by promoting antigen presentation and lymphocyte responses and inducing chemokine expression (Fig. 5a). In addition, myeloid cells in several autoimmune diseases, including SLE, express increased levels of STAT1, suggestive of the priming mechanisms described above that augment IFN responses117. Thus, IFN-mediated priming can contribute to increased cell activation and responsiveness to inflammatory factors in autoimmune diseases. However, IFNs can also have a protective role in autoimmune diseases118. An allelic variant of tyrosine-protein phosphatase non-receptor type 22 (PTPN22) that has been associated with the development of autoimmunity in humans and mouse models was recently shown to confer diminished type I IFN production119, which is consistent with a protective role for type I IFNs. In addition, type I IFNs are protective in animal models of arthritis and inflammatory bowel disease and are therapeutically effective in multiple sclerosis. They are likely to mediate protection in these situations by suppressing the production and function of inflammatory cytokines, suppressing the proliferation of pathogenic cell types, suppressing TH17 cell responses and regulating the blood–brain barrier120. In summary, type I IFNs can have both deleterious and protective effects in autoimmune diseases (Fig. 5a). Their role in human autoimmune diseases will only be clarified with the completion of on-going clinical trials involving IFN blockade. Given that autoimmune diseases are characterized by fluctuating expression of many of the factors described above that regulate type I IFN responses, such as PRR ligands and cytokines, it is likely that type I IFN responses change over time and might predominantly activate or suppress immune responses at different stages of disease.

a | In autoimmune diseases such as systemic lupus erythematosus (SLE), chronic type I interferon (IFN) production is perpetuated in part by dysregulated or persistent stimulation of antigen-presenting cells (APCs), including plasmacytoid dendritic cells (pDCs), by immune complexes and by damage-associated molecular pattern molecules (DAMPs). Persistent type I IFN exposure results in increased T and B cell effector function, leading to autoantibody production and ultimately autoimmune disease. Chronic stimulation by immune complexes and DAMPs (and potentially IFNs) also increases monocyte production of interleukin-10 (IL-10), which suppresses innate and adaptive immunity. Type I IFNs can also suppress cytokine production by innate immune cells. Although type I IFNs are generally thought to promote some autoimmune diseases, such as SLE, their balancing suppressive role might be dominant in other autoimmune diseases such as multiple sclerosis. The immunosuppressive properties of type I IFNs might contribute to increased susceptibility to particular infections in SLE. b | During chronic viral or bacterial infections, cells continuously produce type I IFNs. Chronic type I IFN exposure of innate immune cells ultimately induces immunosuppressive pathways involving IL-10 and programmed cell death 1 ligand 1 (PDL1), which suppress the function of T cells and feed back to also suppress innate immune cells. In chronic lymphocytic choriomeningitis virus (LCMV) and Mycobacterium leprae infections, persistent type I IFN exposure suppresses effective pathogen clearance pathways involving IFNγ. IFNR, IFN receptor; PD1, programmed cell death 1.
Cross-regulation between TNF and IFN signalling pathways. Cross-regulation among type I IFNs and pro-inflammatory cytokines that drive disease pathogenesis is likely to help shape the role of type I IFNs in autoimmune diseases. For example, there is substantial crosstalk between the signalling pathways induced by type I IFNs and TNF, a cytokine that is pathogenic in human autoimmune diseases, including rheumatoid arthritis and inflammatory bowel disease121. Based on findings that type I IFNs and TNF can mutually suppress each other's expression, that TNF blockade can elicit a type I IFN signature in patients' blood and that TNF production is low in the NZB/W mouse model of SLE, Banchereau and colleagues proposed that an imbalance between these two cytokines leads to unrestrained excessive action of TNF or type I IFN, which results in distinct phenotypes of autoimmune disease122. We have discovered additional complexity in the cross-regulation of TNF and type I IFN responses, as we have found that TNF drives ISG expression in synovial macrophages from patients with rheumatoid arthritis123, but at the same time it limits type I IFN-mediated signalling and modulates the pattern of ISG expression (L.T.D., G. Kalliolias and L.B.I., unpublished results). Thus, the regulation of type I IFN responses by TNF might determine the balance between potential pathogenic and protective roles for type IFNs in the pathogenesis of rheumatoid arthritis.
Immunosuppression mediated by type I IFNs. Several recent studies in mice and humans have shown that type I IFNs can have a predominantly suppressive role in the chronic phase of infection with LCMV, Mycobacterium tuberculosis and Mycobacterium leprae114,115,116,126,125,126 (Fig. 5b). During chronic infection of mice with the clone 13 strain of LCMV, type I IFNs induce suppressive factors, such as IL-10 and programmed cell death 1 ligand 1 (PDL1), in addition to ISGs and pro-inflammatory cytokines. The suppressive factors are dominant, as type I IFN blockade or IFNAR1 deficiency results in decreased expression of PDL1 and IL-10 and the concomitant clearance of viral infection, which is dependent on CD4+ T cells and increased IFNγ production. Augmented IL-10 production mediated by type I IFNs also contributes to the exacerbation of disease in a mouse model of M. tuberculosis infection115. Type I IFN-induced IL-10 can suppress M. tuberculosis infection in part by inhibiting production of IL-1, which is important for controlling infection by this pathogen in mice127. Similarly, type I IFNs have a predominantly suppressive role in chronic disseminated lepromatous leprosy lesions in patients infected with M. leprae113. Immunosuppressive mechanisms include the induction of IL-10, which antagonizes the IFNγ-mediated antimicrobial programme that drives pathogen clearance and disease resolution in self-healing tuberculoid lesions113. Anti-proliferative effects of type I IFNs might also contribute to immunosuppression in chronic infections. Collectively, these studies suggest that, when viral or bacterial infections cannot be cleared, sustained type I IFN signalling assumes a predominantly immunosuppressive role, possibly to limit host toxicity and thus limit morbidity during persistent infection. This work has important implications for the role of type I IFNs in chronic human infections such as HIV, HCV and M. tuberculosis. Heightened type I IFN signatures correlate with severity of HCV128,129 and tuberculosis114, which is consistent with an immunosuppressive role for type I IFNs, and increased levels of IL-10 and/or PDL1 have been detected in patients with these infections and in patients with HIV130. Thus, type I IFN-induced immunosuppressive mechanisms might be broadly operative in various chronic infections. These studies suggest a rethinking of not only the role of type I IFNs in chronic infection but also the apparently paradoxical notion that type I IFN-blocking therapies might actually enhance specific immunity and promote clearance of chronic infections.
The aforementioned studies show how the functions of type I IFNs can switch from being primarily antimicrobial and immune-stimulatory in nature during acute infection to being predominantly immunosuppressive at later, chronic stages of infection. It is possible that this switch in function is mediated by some of the mechanisms we have described in this Review that regulate type I IFN responses. This might occur by the unmasking of latent suppressive signalling pathways induced by IFNs. For example, the induction of IL-10 and PDL1 is mediated at least in part by STAT3, and a release of type I IFN-activated STAT3 from repression by SIN3A could help to drive IL-10 and PDL1 expression. An interesting area for future investigation will be an analysis of how type I IFN responses are regulated and reprogrammed during chronic infection and autoimmune diseases, which could be mediated by the epigenetic mechanisms described above.
Closing remarks
Similarly to most cytokines, type I IFNs induce balanced responses in which activating signals that induce antiviral states and promote immune responses are counterbalanced by suppressive signals that limit toxicity to the host and enable coexistence with chronic pathogens. These balanced responses are fine-tuned by host factors at multiple levels, including signalling, transcription and translation, to sculpt immune responses that are appropriate for host defence and survival. Pathogens can exploit some of these regulatory mechanisms to escape immune responses, including the targeting of type I IFN responses by pathogen-encoded molecules131. In addition, dysregulation of type I IFN responses can contribute to autoimmune diseases. A greater understanding of the mechanisms that regulate type I IFN responses is required to guide the development of rational therapies that promote the eradication of pathogens and alleviate autoimmune diseases. This is particularly important for autoimmune diseases in which type I IFNs might be pathological, such as SLE, and also for chronic infections in which unexpected immunosuppressive roles for type I IFNs have recently been described, including infections with HIV, M. leprae and M. tuberculosis that exert an enormous burden on human health. Promising and exciting new areas for further research include the roles of epigenetics, non-coding RNAs, translational regulation and cell metabolism in modulating type I IFN responses. Advances in these areas would aid the development of therapeutics that could selectively target pathogenic components of type I IFN responses while leaving IFN-mediated host defences intact. In addition, new technologies, including those that enable genome-wide analyses, offer promise for a more comprehensive investigation of type I IFN responses in the human immune system110,132,133 and should facilitate the development of targeted therapies.
References
Trinchieri, G. Type I interferon: friend or foe? J. Exp. Med. 207, 2053–2063 (2010).
Pestka, S., Krause, C. D. & Walter, M. R. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 202, 8–32 (2004).
Hertzog, P. J. & Williams, B. R. Fine tuning type I interferon responses. Cytokine Growth Factor Rev. 24, 217–225 (2013).
Paludan, S. R. & Bowie, A. G. Immune sensing of DNA. Immunity 38, 870–880 (2013).
Goubau, D., Deddouche, S. & Reis, E. S. C. Cytosolic sensing of viruses. Immunity 38, 855–869 (2013).
Iwasaki, A. A virological view of innate immune recognition. Annu. Rev. Microbiol. 66, 177–196 (2012).
Levy, D. E. & Darnell, J. E. Jr. STATs: transcriptional control and biological impact. Nature Rev. Mol. Cell Biol. 3, 651–662 (2002).
Stark, G. R. & Darnell, J. E. Jr. The JAK-STAT pathway at twenty. Immunity 36, 503–514 (2012).
MacMicking, J. D. Interferon-inducible effector mechanisms in cell-autonomous immunity. Nature Rev. Immunol. 12, 367–382 (2012).
Schoggins, J. W. et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472, 481–485 (2011). This study identifies specific antiviral functions for multiple ISGs, showing that unique sets of ISGs target distinct viruses. It highlights the importance of translational regulation.
Rusinova, I. et al. Interferome v2.0: an updated database of annotated interferon-regulated genes. Nucleic Acids Res. 41, D1040–D1046 (2012).
Saka, H. A. & Valdivia, R. Emerging roles for lipid droplets in immunity and host-pathogen interactions. Annu. Rev. Cell Dev. Biol. 28, 411–437 (2012).
van Boxel-Dezaire, A. H., Rani, M. R. & Stark, G. R. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity 25, 361–372 (2006).
Gough, D. J., Messina, N. L., Clarke, C. J., Johnstone, R. W. & Levy, D. E. Constitutive type I interferon modulates homeostatic balance through tonic signaling. Immunity 36, 166–174 (2012).
Abt, M. C. et al. Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity 37, 158–170 (2012). Together with references 16 and 17, this study demonstrates that the commensal microflora calibrates innate immune responses and maintains homeostasis in part by providing tonic signals that maintain a basal systemic IFN response.
Ganal, S. C. et al. Priming of natural killer cells by nonmucosal mononuclear phagocytes requires instructive signals from commensal microbiota. Immunity 37, 171–186 (2012).
Kawashima, T. et al. Double-stranded RNA of intestinal commensal but not pathogenic bacteria triggers production of protective interferon-β. Immunity 38, 1187–1197 (2013).
Wang, L. et al. 'Tuning' of type I interferon-induced Jak-STAT1 signaling by calcium-dependent kinases in macrophages. Nature Immunol. 9, 186–193 (2008). This study demonstrates cross-regulation of type I IFN signalling by ITAM-associated receptors, with resultant fine-tuning of ISG induction.
Gilchrist, D. A. et al. Regulating the regulators: the pervasive effects of Pol II pausing on stimulus-responsive gene networks. Genes Dev. 26, 933–944 (2012).
Papadopoulou, A. S. et al. The thymic epithelial microRNA network elevates the threshold for infection-associated thymic involution via miR-29a mediated suppression of the IFN-α receptor. Nature Immunol. 13, 181–187 (2012).
Lu, L. F. et al. Function of miR-146a in controlling Treg cell-mediated regulation of Th1 responses. Cell 142, 914–929 (2010).
Gracias, D. T. et al. The microRNA miR-155 controls CD8+ T cell responses by regulating interferon signaling. Nature Immunol. 14, 593–602 (2013). This study establishes that miRNAs regulate type I IFN responses by demonstrating that miR-155 targets IFN signalling components.
David, M. Interferons and microRNAs. J. Interferon Cytokine Res. 30, 825–828 (2010).
Levy, D. E., Lew, D. J., Decker, T., Kessler, D. S. & Darnell, J. E. Jr. Synergistic interaction between interferon-α and interferon-γ through induced synthesis of one subunit of the transcription factor ISGF3. EMBO J. 9, 1105–1111 (1990). Together with reference 25, this study demonstrates that the priming of augmented IFN responses is mediated by increased expression of IRF9 and STAT1.
Hu, X. et al. Sensitization of IFN-γ Jak-STAT signaling during macrophage activation. Nature Immunol. 3, 859–866 (2002).
Tassiulas, I. et al. Amplification of IFN-α-induced STAT1 activation and inflammatory function by Syk and ITAM-containing adaptors. Nature Immunol. 5, 1181–1189 (2004).
Hu, X., Park-Min, K. H., Ho, H. H. & Ivashkiv, L. B. IFN-γ-primed macrophages exhibit increased CCR2-dependent migration and altered IFN-γ responses mediated by Stat1. J. Immunol. 175, 3637–3647 (2005).
Yarilina, A., Park-Min, K.-H., Antoniv, T., Hu, X. & Ivashkiv, L. B. TNF activates an IRF1-dependent autocrine loop leading to sustained expression of chemokines and STAT1-dependent type I interferon-response genes. Nature Immunol. 9, 378–387 (2008).
Venkatesh, D. et al. Endothelial TNF receptor 2 induces IRF1 transcription factor-dependent interferon-beta autocrine signaling to promote monocyte recruitment. Immunity 38, 1025–1037 (2013).
Cheon, H. et al. IFNβ-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. EMBO J. 32, 2751–2763 (2013).
Fuchs, S. Y. Hope and fear for interferon: the receptor-centric outlook on the future of interferon therapy. J. Interferon Cytokine Res. 33, 211–225 (2013).
de Weerd, N. A. & Nguyen, T. The interferons and their receptors--distribution and regulation. Immunol. Cell Biol. 90, 483–491 (2012).
Bhattacharya, S. et al. Anti-tumorigenic effects of Type 1 interferon are subdued by integrated stress responses. Oncogene 32, 4214–4221 (2013).
Huynh, L., Wang, L., Shi, C., Park-Min, K. H. & Ivashkiv, L. B. ITAM-coupled receptors inhibit IFNAR signaling and alter macrophage responses to TLR4 and Listeria monocytogenes. J. Immunol. 188, 3447–3457 (2012).
Huangfu, W.-C. et al. Inflammatory signaling compromises cell responses to interferon alpha. Oncogene 31, 161–172 (2011).
Liu, J. et al. Virus-induced unfolded protein response attenuates antiviral defenses via phosphorylation-dependent degradation of the type I interferon receptor. Cell Host Microbe 5, 72–83 (2009).
Wang, L. et al. Indirect inhibition of Toll-like receptor and type I interferon responses by ITAM-coupled receptors and integrins. Immunity 32, 518–530 (2010).
Du, Z. et al. Inhibition of IFN-α signaling by a PKC- and protein tyrosine phosphatase SHP-2-dependent pathway. Proc. Natl Acad. Sci. USA 102, 10267–10272 (2005).
Yoshimura, A., Naka, T. & Kubo, M. SOCS proteins, cytokine signalling and immune regulation. Nature Rev. Immunol. 7, 454–465 (2007).
Sarasin-Filipowicz, M. et al. Alpha interferon induces long-lasting refractoriness of JAK-STAT signaling in the mouse liver through induction of USP18/UBP43. Mol. Cell. Biol. 29, 4841–4851 (2009).
Nazarov, P. V. et al. Interplay of microRNAs, transcription factors and target genes: linking dynamic expression changes to function. Nucleic Acids Res. 41, 2817–2831 (2013).
Tili, E. et al. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-α stimulation and their possible roles in regulating the response to endotoxin shock. J. Immunol. 179, 5082–5089 (2007).
Murray, P. J. The JAK-STAT signaling pathway: input and output integration. J. Immunol. 178, 2623–2629 (2007).
Ho, H. H. & Ivashkiv, L. B. Role of STAT3 in type I interferon responses. Negative regulation of STAT1-dependent inflammatory gene activation. J. Biol. Chem. 281, 14111–14118 (2006).
Wang, W. B., Levy, D. E. & Lee, C. K. STAT3 negatively regulates type I IFN-mediated antiviral response. J. Immunol. 187, 2578–2585 (2011).
Nguyen, K. B. et al. Critical role for STAT4 activation by type 1 interferons in the interferon-gamma response to viral infection. Science 297, 2063–2066 (2002). This study demonstrates a switch in type I IFN signalling from STAT1 to STAT4 that shapes CD8+ T cell responses during viral infection in vivo.
Gil, M. P. et al. Regulating type 1 IFN effects in CD8 T cells during viral infections: changing STAT4 and STAT1 expression for function. Blood 120, 3718–3728 (2012).
Tenoever, B. R. et al. Multiple functions of the IKK-related kinase IKKε in interferon-mediated antiviral immunity. Science 315, 1274–1278 (2007).
Ng, S. L. et al. IkappaB kinase epsilon (IKKε) regulates the balance between type I and type II interferon responses. Proc. Natl Acad. Sci. USA 108, 21170–21175 (2012).
Robinson, N. et al. Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nature Immunol. 13, 954–962 (2012).
de Weerd, N. A. et al. Structural basis of a unique interferon-β signaling axis mediated via the receptor IFNAR1. Nature Immunol. 14, 901–907 (2013).
Sadzak, I. et al. Recruitment of Stat1 to chromatin is required for interferon-induced serine phosphorylation of Stat1 transactivation domain. Proc. Natl Acad. Sci. USA 105, 8944–8949 (2008).
Bancerek, J. et al. CDK8 kinase phosphorylates transcription factor STAT1 to selectively regulate the interferon response. Immunity 38, 250–262 (2013).
Yang, J. et al. Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proc. Natl Acad. Sci. USA 107, 21499–21504 (2010).
Droescher, M., Begitt, A., Marg, A., Zacharias, M. & Vinkemeier, U. Cytokine-induced paracrystals prolong the activity of signal transducers and activators of transcription (STAT) and provide a model for the regulation of protein solubility by small ubiquitin-like modifier (SUMO). J. Biol. Chem. 286, 18731–18746 (2011).
Hu, X. & Ivashkiv, L. B. Cross-regulation of signaling pathways by interferon-γ: implications for immune responses and autoimmune diseases. Immunity 31, 539–550 (2009).
Tamura, T., Yanai, H., Savitsky, D. & Taniguchi, T. The IRF family transcription factors in immunity and oncogenesis. Annu. Rev. Immunol. 26, 535–584 (2008).
Levy, D. E., Marie, I., Smith, E. & Prakash, A. Enhancement and diversification of IFN induction by IRF-7-mediated positive feedback. J. Interferon Cytokine Res. 22, 87–93 (2002).
Farlik, M. et al. Contribution of a TANK-binding kinase 1-interferon (IFN) regulatory factor 7 pathway to IFN-γ-induced gene expression. Mol. Cell. Biol. 32, 1032–1043 (2012).
Qiao, Y. et al. Synergistic activation of inflammatory cytokine genes by interferon-γ-induced chromatin remodeling and Toll-like receptor signaling. Immunity 39, 454–469 (2013). Together with references 79 and 80, this study demonstrates a pervasive binding of STAT1 to promoters and enhancers genome-wide that programs cellular responses to environmental cues.
Chatterjee-Kishore, M., Wright, K. L., Ting, J. P. & Stark, G. R. How Stat1 mediates constitutive gene expression: a complex of unphosphorylated Stat1 and IRF1 supports transcription of the LMP2 gene. EMBO J. 19, 4111–4122 (2000).
Farlik, M. et al. Nonconventional initiation complex assembly by STAT and NF-κB transcription factors regulates nitric oxide synthase expression. Immunity 33, 25–34 (2010).
Xu, D. et al. Promyelocytic leukemia zinc finger protein regulates interferon-mediated innate immunity. Immunity 30, 802–816 (2009).
Litvak, V. et al. A FOXO3-IRF7 gene regulatory circuit limits inflammatory sequelae of antiviral responses. Nature 490, 421–425 (2012).
Shalek, A. K. et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature 498, 236–240 (2013).
Zhao, M., Zhang, J., Phatnani, H., Scheu, S. & Maniatis, T. Stochastic expression of the interferon-beta gene. PLoS Biol. 10, e1001249 (2012).
Hwang, S. Y. et al. Biphasic RLR-IFN-β response controls the balance between antiviral immunity and cell damage. J. Immunol. 190, 1192–1200 (2013).
Bell, O., Tiwari, V. K., Thoma, N. H. & Schubeler, D. Determinants and dynamics of genome accessibility. Nature Rev. Genet. 12, 554–564 (2011).
Smale, S. T. Selective transcription in response to an inflammatory stimulus. Cell 140, 833–844 (2010).
Yan, Z. et al. PBAF chromatin-remodeling complex requires a novel specificity subunit, BAF200, to regulate expression of selective interferon-responsive genes. Genes Dev. 19, 1662–1667 (2005).
Liu, H., Kang, H., Liu, R., Chen, X. & Zhao, K. Maximal induction of a subset of interferon target genes requires the chromatin-remodeling activity of the BAF complex. Mol. Cell. Biol. 22, 6471–6479 (2002).
Cui, K. et al. The chromatin-remodeling BAF complex mediates cellular antiviral activities by promoter priming. Mol. Cell. Biol. 24, 4476–4486 (2004).
Huang, M. et al. Chromatin-remodelling factor BRG1 selectively activates a subset of interferon-α-inducible genes. Nature Cell Biol. 4, 774–781 (2002).
Ni, Z. et al. Apical role for BRG1 in cytokine-induced promoter assembly. Proc. Natl Acad. Sci. USA 102, 14611–14616 (2005).
Ramirez-Carrozzi, V. R. et al. Selective and antagonistic functions of SWI/SNF and Mi-2β nucleosome remodeling complexes during an inflammatory response. Genes Dev. 20, 282–296 (2006). This study establishes the importance of chromatin remodelling for the induction of inflammatory genes and ISGs.
Ramirez-Carrozzi, V. R. et al. A unifying model for the selective regulation of inducible transcription by CpG islands and nucleosome remodeling. Cell 138, 114–128 (2009).
Bhatt, D. M. et al. Transcript dynamics of proinflammatory genes revealed by sequence analysis of subcellular RNA fractions. Cell 150, 279–290 (2012).
Zentner, G. E. & Henikoff, S. Regulation of nucleosome dynamics by histone modifications. Nature Struct. Mol. Biol. 20, 259–266 (2013).
Vahedi, G. et al. STATs shape the active enhancer landscape of T cell populations. Cell 151, 981–993 (2012).
Ostuni, R. et al. Latent enhancers activated by stimulation in differentiated cells. Cell 152, 157–171 (2013).
Hargreaves, D. C., Horng, T. & Medzhitov, R. Control of inducible gene expression by signal-dependent transcriptional elongation. Cell 138, 129–145 (2009).
Nicodeme, E. et al. Suppression of inflammation by a synthetic histone mimic. Nature 468, 1119–1123 (2010). This study demonstrates the feasibility of therapeutic targeting of chromatin regulatory proteins to selectively suppress inflammatory gene and ISG expression.
Patel, M. C. et al. BRD4 coordinates recruitment of pause release factor P-TEFb and the pausing complex NELF/DSIF to regulate transcription elongation of interferon-stimulated genes. Mol. Cell. Biol. 33, 2497–2507 (2013).
Shakespear, M. R., Halili, M. A., Irvine, K. M., Fairlie, D. P. & Sweet, M. J. Histone deacetylases as regulators of inflammation and immunity. Trends Immunol. 32, 335–343 (2011).
Fonseca, G. J. et al. Adenovirus evasion of interferon-mediated innate immunity by direct antagonism of a cellular histone posttranslational modification. Cell Host Microbe 11, 597–606 (2012).
Lau, J. F., Nusinzon, I., Burakov, D., Freedman, L. P. & Horvath, C. M. Role of metazoan mediator proteins in interferon-responsive transcription. Mol. Cell. Biol. 23, 620–628 (2003).
Gnatovskiy, L., Mita, P. & Levy, D. E. The human RVB complex is required for efficient transcription of type I IFN-stimulated genes. Mol Cell. Biol. 33, 3817–3825 (2013).
Flammer, J. R. et al. The type I interferon signaling pathway is a target for glucocorticoid inhibition. Mol. Cell. Biol. 30, 4564–4574 (2010).
Icardi, L. et al. The Sin3a repressor complex is a master regulator of STAT transcriptional activity. Proc. Natl Acad. Sci. USA 109, 12058–12063 (2012). This study reveals the association of co-repressors with specific STATs as a mechanism that can selectively silence the expression of subsets of type I IFN response genes.
Tahk, S. et al. Control of specificity and magnitude of NF-κB and STAT1-mediated gene activation through PIASy and PIAS1 cooperation. Proc. Natl Acad. Sci. USA 104, 11643–11648 (2007).
Shuai, K. & Liu, B. Regulation of gene-activation pathways by PIAS proteins in the immune system. Nature Rev. Immunol. 5, 593–605 (2005).
Liu, B., Tahk, S., Yee, K. M., Fan, G. & Shuai, K. The ligase PIAS1 restricts natural regulatory T cell differentiation by epigenetic repression. Science 330, 521–525 (2010).
Marazzi, I. et al. Suppression of the antiviral response by an influenza histone mimic. Nature 483, 428–433 (2012).
Natoli, G., Ghisletti, S. & Barozzi, I. The genomic landscapes of inflammation. Genes Dev. 25, 101–106 (2011).
Fang, T. C. et al. Histone H3 lysine 9 di-methylation as an epigenetic signature of the interferon response. J. Exp. Med. 209, 661–669 (2012).
Ea, C. K., Hao, S., Yeo, K. S. & Baltimore, D. EHMT1 protein binds to nuclear factor-κB p50 and represses gene expression. J. Biol. Chem. 287, 31207–31217 (2012).
Walsh, D., Mathews, M. B. & Mohr, I. Tinkering with translation: protein synthesis in virus-infected cells. Cold Spring Harb. Perspect. Biol. 5, a012351 (2013).
Kaur, S. et al. Regulatory effects of mTORC2 complexes in type I IFN signaling and in the generation of IFN responses. Proc. Natl Acad. Sci. USA 109, 7723–7728 (2012).
Kaur, S. et al. Role of the Akt pathway in mRNA translation of interferon-stimulated genes. Proc. Natl Acad. Sci. USA 105, 4808–4813 (2008).
Joshi, S., Kaur, S., Kroczynska, B. & Platanias, L. C. Mechanisms of mRNA translation of interferon stimulated genes. Cytokine 52, 123–127 (2010).
Rani, M. R., Hibbert, L., Sizemore, N., Stark, G. R. & Ransohoff, R. M. Requirement of phosphoinositide 3-kinase and Akt for interferon-β-mediated induction of the beta-R1 (SCYB11) gene. J. Biol. Chem. 277, 38456–38461 (2002).
Thoreen, C. C. et al. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 485, 109–113 (2012).
Hsieh, A. C. et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 485, 55–61 (2012).
Colina, R. et al. Translational control of the innate immune response through IRF-7. Nature 452, 323–328 (2008).
Ruggieri, A. et al. Dynamic oscillation of translation and stress granule formation mark the cellular response to virus infection. Cell Host Microbe 12, 71–85 (2012).
Terenzi, F., Hui, D. J., Merrick, W. C. & Sen, G. C. Distinct induction patterns and functions of two closely related interferon-inducible human genes, ISG54 and ISG56. J. Biol. Chem. 281, 34064–34071 (2006).
Fensterl, V. & Sen, G. C. The ISG56/IFIT1 gene family. J. Interferon Cytokine Res. 31, 71–78 (2011).
Lee, M. S., Kim, B., Oh, G. T. & Kim, Y. J. OASL1 inhibits translation of the type I interferon-regulating transcription factor IRF7. Nature Immunol. 14, 346–355 (2013).
Hall, J. C. & Rosen, A. Type I interferons: crucial participants in disease amplification in autoimmunity. Nature Rev. Rheumatol. 6, 40–49 (2010).
Maurano, M. T. et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195 (2012).
Rai, E. & Wakeland, E. K. Genetic predisposition to autoimmunity—what have we learned? Semin. Immunol. 23, 67–83 (2011).
Forster, S. Interferon signatures in immune disorders and disease. Immunol. Cell Biol. 90, 520–527 (2012).
Teles, R. M. B. et al. Type I interferon suppresses type II interferon-triggered human anti-mycobacterial responses. Science 339, 1448–1453 (2013). Together with references 114–116 and 124–127, this study demonstrates a dominant suppressive function of type I IFNs in chronic infections that is mediated by the induction of IL-10 and PDL1.
Berry, M. P. et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 466, 973–977 (2010).
McNab, F. W. et al. TPL-2-ERK1/2 signaling promotes host resistance against intracellular bacterial infection by negative regulation of type I IFN production. J. Immunol. 191, 1732–1743 (2013).
O'Garra, A. et al. The immune response in tuberculosis. Annu. Rev. Immunol. 31, 475–527 (2013).
Kalliolias, G. D. & Ivashkiv, L. B. Overview of the biology of type I interferons. Arthritis Res. Ther. 12 (Suppl. 1), S1 (2010).
Ivashkiv, L. B. PTPN22 in autoimmunity: different cell and different way. Immunity 39, 91–93 (2013).
Wang, Y. et al. The autoimmunity-associated gene PTPN22 potentiates toll-like receptor-driven, type 1 interferon-dependent immunity. Immunity 39, 111–122 (2013).
Prinz, M. et al. Distinct and nonredundant in vivo functions of IFNAR on myeloid cells limit autoimmunity in the central nervous system. Immunity 28, 675–686 (2008).
Cantaert, T., Baeten, D., Tak, P. P. & van Baarsen, L. G. Type I IFN and TNFalpha cross-regulation in immune- mediated inflammatory disease: basic concepts and clinical relevance. Arthritis Res. Ther. 12, 219 (2010).
Banchereau, J., Pascual, V. & Palucka, A. K. Autoimmunity through cytokine-induced dendritic cell activation. Immunity 20, 539–550 (2004).
Gordon, R. A., Grigoriev, G., Lee, A., Kalliolias, G. D. & Ivashkiv, L. B. The interferon signature and STAT1 expression in rheumatoid arthritis synovial fluid macrophages are induced by tumor necrosis factor alpha and counter-regulated by the synovial fluid microenvironment. Arthritis Rheum. 64, 3119–3128 (2012).
Wilson, E. B. et al. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 340, 202–207 (2013).
Teijaro, J. R. et al. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 340, 207–211 (2013).
Antonelli, L. R. et al. Intranasal Poly-IC treatment exacerbates tuberculosis in mice through the pulmonary recruitment of a pathogen-permissive monocyte/macrophage population. J. Clin. Invest. 120, 1674–1682 (2010).
Mayer-Barber, K. D. et al. Innate and adaptive interferons suppress IL-1α and IL-1β production by distinct pulmonary myeloid subsets during Mycobacterium tuberculosis infection. Immunity 35, 1023–1034 (2011).
Su, A. I. et al. Genomic analysis of the host response to hepatitis C virus infection. Proc. Natl Acad. Sci. USA 99, 15669–15674 (2002).
Guidotti, L. G. & Chisari, F. V. Immunobiology and pathogenesis of viral hepatitis. Annu. Rev. Pathol. 1, 23–61 (2006).
Kulpa, D. A. et al. PD-1 coinhibitory signals: the link between pathogenesis and protection. Semin. Immunol. 25, 219–227 (2013).
Hajishengallis, G. & Lambris, J. D. Microbial manipulation of receptor crosstalk in innate immunity. Nature Rev. Immunol. 11, 187–200 (2011).
Maecker, H. T., McCoy, J. P. & Nussenblatt, R. Standardizing immunophenotyping for the Human Immunology Project. Nature Rev. Immunol. 12, 191–200 (2012).
Davis, M. M. Immunology taught by humans. Sci Transl. Med. 4, 117fs2 (2012).
Fung, K. Y. et al. Interferon-ε protects the female reproductive tract from viral and bacterial infection. Science 339, 1088–1092 (2013).
Wang, H. et al. The role of glycogen synthase kinase 3 in regulating IFN-β-mediated IL-10 production. J. Immunol. 186, 675–684 (2011).
Chen, X. et al. Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc. Natl Acad. Sci. USA 109, E2865–E2874 (2012).
Khabar, K. S. & Young, H. A. Post-transcriptional control of the interferon system. Biochimie 89, 761–769 (2007).
Schulz, O. et al. Protein kinase R contributes to immunity against specific viruses by regulating interferon mRNA integrity. Cell Host Microbe 7, 354–361 (2010).
Acknowledgements
We thank P. Crow for critical review of the manuscript. This work was supported by grants from the US National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Glossary
- MicroRNAs
-
(miRNAs). Small RNA molecules that regulate the expression of genes by binding to the 3′ untranslated regions of specific mRNAs.
- Degron sequence
-
Degrons are signals within proteins that target them for rapid degradation. Degrons can be overt, as in the case of the N-end rule, or covert, as in the case of cyclins. For example, cyclin B must be rapidly destroyed after mitosis, and this is achieved by kinase-regulated access to a 'destruction box' sequence in cyclin B that stimulates polyubiquitylation and subsequent degradation by the proteasome.
- Mediator
-
A multiprotein complex that functions as a co-activator of transcription in all eukaryotes.
- Sumoylated
-
A sumoylated protein has undergone a type of post-translational protein modification in which the ubiquitin-like protein SUMO (small ubiquitin-related modifier) is covalently attached to the protein by an enzymatic cascade that is analogous to the cascade involved in protein ubiquitylation.
- CpG islands
-
Sequences of 0.5–2 kilobases that are rich in CpG dinucleotides. They are mostly located upstream of housekeeping genes and also of some tissue-specific genes. They are constitutively hypomethylated in many animal cell types.
- RVB proteins
-
ATP-binding proteins that belong to the ATPase- associated with diverse cellular activities (AAA) family of ATPases. They are found in different protein and nucleoprotein complexes that have roles in diverse cellular responses, including transcription, mitosis, development, apoptosis and DNA damage responses.
- IFN signature
-
A pattern of increased expression of interferon- stimulated genes (ISGs) in tissue samples or stimulated cells. The IFN signature is typically detected by using a high-throughput approach, such as microarray or RNA sequencing, to analyse gene expression.
Rights and permissions
About this article
Cite this article
Ivashkiv, L., Donlin, L. Regulation of type I interferon responses. Nat Rev Immunol 14, 36–49 (2014). https://doi.org/10.1038/nri3581
Published:
Issue Date:
DOI: https://doi.org/10.1038/nri3581
This article is cited by
-
Role of sex in immune response and epigenetic mechanisms
Epigenetics & Chromatin (2024)
-
The helicase domain of human Dicer prevents RNAi-independent activation of antiviral and inflammatory pathways
The EMBO Journal (2024)
-
Role of MDA5 Deficiency in Pathogenesis of Fungal Infections
Indian Journal of Pediatrics (2024)
-
Personalizing Oncolytic Immunovirotherapy Approaches
Molecular Diagnosis & Therapy (2024)
-
Evolutionary and functional conservation of IRF7 in the Tibetan frog Nanorana parkeri
Molecular Biology Reports (2024)
