
Abstract
Accumulation of mutations within the genome is the primary driving force in viral evolution within an endemic setting. This inherent feature often leads to altered virulence, infectivity and transmissibility, and antigenic shifts to escape host immunity, which might compromise the efficacy of vaccines and antiviral drugs. Therefore, we carried out a genome-wide analysis of circulating SARS-CoV-2 strains to detect the emergence of novel co-existing mutations and trace their geographical distribution within India. Comprehensive analysis of whole genome sequences of 837 Indian SARS-CoV-2 strains revealed the occurrence of 33 different mutations, 18 of which were unique to India. Novel mutations were observed in the S glycoprotein (6/33), NSP3 (5/33), RdRp/NSP12 (4/33), NSP2 (2/33), and N (1/33). Non-synonymous mutations were found to be 3.07 times more prevalent than synonymous mutations. We classified the Indian isolates into 22 groups based on their co-existing mutations. Phylogenetic analysis revealed that the representative strains of each group were divided into various sub-clades within their respective clades, based on the presence of unique co-existing mutations. The A2a clade was found to be dominant in India (71.34%), followed by A3 (23.29%) and B (5.36%), but a heterogeneous distribution was observed among various geographical regions. The A2a clade was highly predominant in East India, Western India, and Central India, whereas the A2a and A3 clades were nearly equal in prevalence in South and North India. This study highlights the divergent evolution of SARS-CoV-2 strains and co-circulation of multiple clades in India. Monitoring of the emerging mutations will pave the way for vaccine formulation and the design of antiviral drugs.
Similar content being viewed by others

Introduction
When a virus adapts to a new host within an endemic setting, it needs to exploit the host’s cellular machinery for successful entry, establishing its replication, and evading the host’s immune responses [1]. To achieve this, viruses modify antigenic epitopes on their proteins by continuously mutating their genomes. If the virus evolves in a stable environment with minimal selection, transition mutations are more frequent than the transversions [2]. Accumulation of deleterious mutations, which may include insertion, deletion, or substitution mutations, are filtered out through natural selection, either by reverting back to the ancestral state or by getting fixed with compensatory mutations that offset the effects of deleterious mutations while advantageous mutations persist [2,3,4,5]. Hence, digging deep into the type of mutations that occur may help in understanding how selection pressure might be acting on a novel virus [6].
In pursuit of the origin of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), researchers found traces of its zoonotic transmission, as a number of the initial cases were reported in people visiting the Wuhan Seafood Market [7,8,9]. The transmission dynamics of this virus were a major focus of research in the early period of the pandemic, where there were numerous controversies and questions. Phylogenetic analysis of the virus isolated from infected individuals revealed a high degree of sequence similarity to bat-infecting SARS-CoV strains in the subgenus Sarbecovirus of the family Coronaviridae [10]. Later on, when it was found that patient zero, along with a few subsequent emerging cases of this disease, did not share any history of exposure to the seafood market, it was concluded upon further investigations that Malayan pangolins might have served as intermediate hosts for SARS-CoV-2 before it could reach its pinnacle host, humans [11, 12]. RNA viruses possess the characteristic feature of high mutability, and SARS-CoV-2, a positive-strand RNA virus, has been evolving at a rapid rate since its emergence in Wuhan at the end of 2019 [13,14,15]. In India, the first case was registered on January 30, 2020, and by the month of August, the number of registered cases of infection had reached 3,542,733 with the total number of deaths reaching 63,498 [16]. An accurate determination of the number of cases is difficult because of the high percentage of SARS-CoV-2-infected asymptomatic carriers [17]. However, in the span of six months (February 2020 to August 2020), the circulating SARS-CoV-2 strains in India accumulated a large number of mutations, which might have resulted in altered virulence, infectivity, or transmissibility [18, 19]. The evolution of viruses frequently relies on the co-occurrence of multiple mutations in different genes or within a single gene. The accumulation of mutations in viruses such as influenza virus has been shown to result in emergence of vaccine escape mutants or drug-resistant mutants, leading to a continuous need to develop new vaccines or drugs [20]. Continuous monitoring of single-nucleotide polymorphisms and locating them in protein-coding genes might help in gaining insight into the genetic diversity and evolution of SARS-CoV-2, which is also important for designing effective vaccines or antiviral drugs against this virus [21].
This study was designed to extensively analyze and compare genetic mutations in SARS-CoV-2 strains from various geographical regions in India, using the prototype ‘Wuhan strain’ as a reference [22]. Establishing an atlas of co-existing mutations in the SARS-CoV-2 genome might help to trace their genetic evolution and address the relationship between the type and strength of co-existing mutations with the rate of adaptation within the various epidemiological settings of India.
Materials and methods
Sequence retrieval
Full genome nucleotide sequences of 837 SARS-CoV-2 viruses circulating in India in March-August 2020 were retrieved from the GISAID repository [23] (Supplementary Table I). Several other clade-specific reference genome sequences of SARS-CoV-2 were also downloaded from GISAID for construction of the dendrogram.
Screening of mutations and phylogenetic analysis
Novel mutations in the genome of Indian SARS-CoV-2 isolates were analysed with respect to the prototype strain Wuhan-Hu-1 (GenBank MN908947.3). A phylogenetic dendrogram was constructed based on the complete genome sequences of 22 representative Indian SARS-CoV-2 strains and 10 reference SARS-CoV-2 strains, using Molecular Evolutionary Genetics Analysis (MEGA) version X [24], using general time-reversible model as the the maximum-likelihood statistical method with 500 bootstrap replicates, using the best-fit nucleotide substitution model. MUSCLE v3.8.31 was used for multiple sequence alignment [25]. Amino acid sequences were retrieved using the TRANSEQ nucleotide-to-protein sequence conversion tool (EMBL-EBI, Cambridgeshire, UK) [26].
Results
Identification and analysis of mutations in SARS-CoV-2 strains circulating in different geographical regions of India
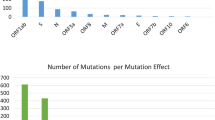
To identify mutations in the SARS-CoV-2 genome accumulating through natural selection, we performed whole-genome sequence analysis of 837 Indian SARS-CoV-2 strains that had been deposited in the GISAID repository (Supplementary Fig. 1). A total of 33 different mutations were found in the 837 Indian isolates in comparison to the prototype strain Wuhan-Hu-1 (only those that were found in at least five isolates were considered). Eight substitution mutations were found in the S protein, six of which were non-synonymous (G21724T/L54F, A21792T/K77M, G21795T/R78M, G23311T/E583D, A23403G/D614G, and G23593T/Q677H) and two were synonymous (C22444T/D294D and C23929T/Y789Y). Seven mutations were found in the NSP3 protein, six of which were non-synonymous (G4866T/G716I, C4965T/T749I, C5700A/A994D, A6081G/D1121G, C6310A/S1197R, and C6312A/T1198K) and one was synonymous (C3037T/F106F). Five non-synonymous mutations were found in RdRp/NSP12 (C13730T/A97V, C14408T/P323L, C14425A/L329I, G15451A/G571S, G16078A/V880I), four in NSP2 (C884T/R27C, G1397A/V198I, C1707T/S301F, G1820A/G339S), three in N (T28311C/P13L, C28854T/S194L, GGG28881AAC/RG203KR) and two in NSP4 (G8653T/M33I, C8782T/S76S). Single mutations were identified in the 5’-UTR region (C241T), NSP6 (G11083T/L37F), ORF3a (G25563T/Q57H), and ORF8 (T28144C/L84S). The rest of the genome was found to be conserved, with no significant amino acid substitutions. The S, NSP3, and NSP12 proteins were found to be the most susceptible to mutations, followed by NSP2, N, NSP4, NSP6, ORF3a and ORF8 (Fig. 1A). Four mutations, C241T in the 5’-UTR (n = 589/837), C3037T/F106F in NSP3 (n = 588/837), C14408T/P323L in RdRP (n = 587/837), and A23403G/D614G in S (n = 586/837) were found to predominate in all geographic regions of India (Fig. 1B-G). G25563T/Q57H in ORF3a (n = 190/837) was the next most frequent mutation in India, principally in Western India. Other frequent mutations included G11083T/L37F in NSP6 (n = 189/837), C13730T/A97V in RdRP (n = 182/837), C23929T/Y789Y in S (n = 176/837), T28311C/P13L in N (n = 182/837), and C6312A/T1198K in NSP3 (n = 166/837), which were found primarily in South and North India (Fig. 1B-G). These mutations were followed in frequency by GGG28881AAC/RG203KR in N (n = 163/837, mostly in South India), C22444T/D294D in S (n = 118/837, mostly in Western India), C28854T/S194L in N (n = 117/837, mostly in Western India), C5700A/A994D in NSP3 (n = 82/837, mostly in Western India), T28144C/L84S in ORF8 (n = 44/837, mostly in East India), C8782T/S76S in NSP4 (n = 44/837, mostly in East India), C6310A/S1197R in NSP3 (n = 35/837, mostly in East India), and G21724T/L54F in S (n = 21/837, mostly in Western India) (Fig. 1B-G). The most amino acid sequence variation was observed in the NSP3, NSP4, RdRp, S, and N proteins of strains circulating in Western India (Fig. 1D), whereas North India had the most mutations in NSP2 (Fig. 1G).

(A-B): Identification of various mutations present in the genome of SARS-CoV-2 circulating in India. (A) Pictorial representation of 33 different mutations (at both the nucleotide and amino acid levels) found in different regions (coding and non-coding regions) of the SARS-CoV-2 genome. (B) Relative frequencies of 33 different mutations in India. (C-G) Identification of various mutations present in the genome of SARS-CoV-2 circulating in different geographic regions in India. Relative frequencies of various mutations in (C) East India, (D) Western India, (E) South India, (F) Central India and (G) North India
Emergence of synonymous and non-synonymous mutations: Analysis of nucleotide substitution events (transition and transversion) at the level of codon positions
Analysis of mutational events per sample revealed that the most frequent number of mutations per genome in Indian isolates was five, followed by four, six, seven and two (Fig. 2A). Non-synonymous mutations occurred 3.07 times (2844/926) more frequently than synonymous mutations (Fig. 2B). We identified eight nucleotide substitutions – four transitions (C>T, A>G, G>A, T>C) and four transversions (G>T, C>A, G>C, A>T) – that were responsible for 29 non-synonymous and four synonymous mutations (Fig. 2C). The C>T transition was the prevalent substitution (Fig. 2C), occurring most frequently in the second position of the codon, followed by the third position (Fig. 2D). C>T transitions resulted in six non-synonymous mutations (A97V, P13L, S194L, S301F, T749I) in the second position of the codon and four synonymous mutations (D294D, Y789Y, F106F, S76S) in the third position (Fig. 2D). The second most frequent substitution was an A>G transition, which occurred solely at the second position of the codon and was responsible for the D614G and D1121G mutations. The G>T transversion was the next most frequent nucleotide substitution, occurring frequently in the third position and rarely in the second position of the codon, generating six (E583D, Q667H, L54F, M33I, L37F, Q57H) and two (R78M, S716I) non-synonymous mutations, respectively. A G>A transition was the next most frequent substitution, occurring either in the first position, leading to V880I, G671S, V198I or G339S, or in both the second and third positions of the codon, resulting in the R203K change. A C>A transversion was seen more frequently in the second position of the codon, generating the T1198K and A994D mutations, although it did appear occasionally in the first position, causing the L329I and S1197R mutations. A G>C transversion occurred only in the first position of the codon that produced the G204R mutation. T>C and A>T were the least frequent substitutions, occurring mostly in the second position of the codon and were responsible for the L84S and K77M mutation, respectively (Fig. 2C-D).

Analysis of synonymous and non-synonymous mutations regarding nucleotide substitutions at different positions in codons. (A) Frequency distribution of SARS-CoV-2 isolates harbouring varying numbers of co-existing mutations. (B) Prevalence of synonymous and non-synonymous mutations in SARS-CoV-2 genomes across India. (C) Frequency distribution of various transitional (C>T, A>G, G>A and T>C) and transversional (G>T, C>A, G>C and A>T) substitution events. (D) Frequency distribution of various types of substitutional events occurring at the first, second, and third nucleotide positions of the codon.
Geographical classification of the Indian SARS-CoV-2 strains based on co-existing mutations and their preponderance in different geographical regions across India
On the basis of co-existing mutations, we could classify the 820 Indian isolates into 22 groups, each group representing a different set of co-existing mutations (Fig. 3A, Table 1). Out of 22 groups, 12 represented the strains belonging to the A2a clade (the most prevalent), with four clade-specific mutations (D614G/S, F106F/NSP3, C241T/5’-UTR and P323L/RdRp). Eleven out of 12 groups have acquired additional mutations (Q57H/ORF3a, S194L/N, D294D/S, V880I/RdRp, E583D/RdRp, L54F/S, R78M/S, RG203KR/N, A994D/NSP3, G671S/RdRp, A97V/RdRp, L291I/RdRp) in various combinations. The group with only four characteristic mutations was the predominant one within the A2a clade and was the largest group in India (Fig. 3A). The groups with the four common mutations along with Q57H; Q57H, S194L and D294D; RG203KR; or RG203KR and A994D were moderately frequent within the A2a clade. Eight groups represented the A3 clade bearing an L37F mutation along with various combinations of V198I/NSP2, M33I/NSP4, R27C/NSP2, P13L/Y789Y/S, A97V/RdRp, T1198K/NSP3, S1197R/NSP3, S301F/NSP2, G339S/NSP2, D1121G/NSP3, and K77M/S. A group with the mutations L37F, P13L, Y789Y, A97V and T1198K was the predominant group in the A3 clade and the second largest group in India. Two groups representing the B clade had L84S/ORF8 and S76S/NSP4 substitutions with or without T749I/NSP3. Overall, the dominant clades in India were A2a (71.34%), followed by A3 (23.29%) and B (5.36%) (Fig. 3B). The A2a clade was predominant across East, West and Central India, whereas the A3 and A2a clades were equally distributed in South and North India. B clade strains were only found in East and West India (Fig. 4A-F).

Grouping of SARS-CoV-2 strains on the basis of co-existing mutations and analysis of their prevalence. (A) Analysis of mutations revealed the presence of the clades (A2a, A3 and B) of SARS-CoV-2 strains in India. The accumulation of novel mutations in addition to clade-specific variations allowed us to classify A2a clade strains into 12 groups, A3 clade strains into eight groups, and B clade strains into two groups. We also show the number of strains belonging to each group. (B) Prevalence of three clade-specific mutations in India. The A2a clade (71.34%) was found to be the most prevalent in India, followed by A3 (23.29%) and B (5.36%).

Prevalence of three different clades (A2a, A3 and B) and their subgroups in different geographic regions in India. (A-C) Frequency distribution of strains belonging to each group of three different clades in (A) East India, (B) Western India, and (C) South India. (D-F): Frequency distribution of strains belonging to each group of three different clades in (D) Central India and (E) North India. (F) Prevalence of three different clades in different geographic regions of India.
Phylogenetic analysis of 22 groups of Indian SARS-CoV-2 isolates in comparison to the various clade-specific strains
The genetic relationships and clustering pattern of the 22 groups of Indian isolates were analyzed by comparing them with SARS-CoV-2 strains representing various clades and the prototype clade O strain from Wuhan (MN908947.3). Whole genome sequences of 22 representative Indian strains (one from each of the 22 co-evolving mutant groups) (Table 1) along with strains representing 10 different clades were selected for phylogenetic analysis. As expected, the dendrogram revealed that the 22 isolates clustered with strains of three different clades (12 strains with A2a, eight with A3 and two with B4-2). The prototype strain (clade O) belonged to the lineage including the A3 and B clade strains. Very interestingly, the 22 Indian strains, representing different groups, generated sub-clusters within their respective clades, based on the accumulation of co-existing mutations in addition to the clade-specific mutations (shown on the branch of each lineage) (Fig. 5, Table 1). Within the A3 clade, two sub-clusters were seen: a (one strain) and b (six strains), bearing three (V198I, M33I, R27C) and at least four (P13L, Y789Y, A97V, T1198K) co-existing mutations, respectively, in addition to the characteristic L37F mutation. Within clade-B4-2, only one representative strain with a T749I mutation in addition to the clade-specific L84S and S76S mutations was observed. Indian strains within clade A2a formed five sub-clusters (viz., a, six strains; b, c, and d, one strain each; and e, two strains). In addition to the four A2a clade-specific mutations (D614G, F106F, C241T and P323L); other novel variations, including Q57H, A97V, S716I and L329I, G671S, and RG203KR, were found in sub-cluster a-e strains. All of the representative Indian strains had >99% nucleotide sequence identity to each other. The prototype strain belonging to the O clade clustered close to the A3 clade (>98% identity).

Molecular phylogenetic analysis by the maximum-likelihood method. The phylogenetic dendrogram is based on whole genome sequences of 22 representative strains from 22 different groups together with representatives of nine clades specific known strains and the prototype O clade strain (MN908947.3). Twenty-two representative strains are indicated by an asterisk (*). The scale bar represents 0.00005 nucleotide substitution per site. Bootstrap values less than 70% are not shown. The best-fit model used for constructing the phylogenetic dendrogram was the general time-reversible model (GTR).
Discussion
During the emergence of SARS-CoV-2 virus in Wuhan, the monophyletic clade O prevailed. As the virus spread across the continents, it started accumulating mutations to adapt in various epidemiological settings. In the present study, we performed a comprehensive mutational analysis of 837 Indian SARS-CoV-2 strains identified in different geographical regions of India and classified them on the basis of co-existing mutations.
Our data highlighted the existence of 33 different mutations (32 mutations in nine different protein coding genes and one in the 5’-UTR) among Indian SARS-CoV-2 strains. The largest number of mutations was detected in the S protein (8) followed by NSP3 (7), NSP12 (5), NSP2 (4), N (3), and NSP4 (2). Only a single mutation was observed in the 5’-UTR and the NSP6, ORF3a and ORF8 genes. Along with 28 non-synonymous mutations, we observed four silent mutations (D294D/S, F106F/NSP3, S76S/NSP4 and Y789Y/S), which may not have any apparent effect on protein structure but may alter codon usage and therefore affect the efficiency of translation [26]. Mutations in the 5’-UTR region can have a significant impact on folding, transcription and replication of the viral genome [26]. Comparing the mutation patterns in India and abroad, we observed certain mutations in S (L54F, K77M, R78M, D294D, E583D, Q677H), NSP3 (G716I, T749I, A994D, D1121G, S1197R), RdRP (A97V, L329I, G571S, V880I), NSP2 (S301F, G339S), and N (S194L) that are unique to Indian isolates, whereas the rest of the 33 mutations had already been reported in other countries [26, 27]. An analysis of missense mutations in 128 SARS-CoV-2-infected Indian patients, mostly from Ahmedabad and Gujarat, had detected some of the mutations that we report here [28]. D614G/S, a characteristic mutation of the A2 clade that was first reported in Germany [29], has been found to correlate strongly with high infectivity [30, 31]. Recent reports have also suggested that residue D614 lies within an immunodominant linear epitope of the S protein and induces an exaggerated serological response. The D614G mutation has also been established to be associated with reduced sensitivity of neutralizing antibodies toward the S protein [31, 32]. Among the five novel non-synonymous mutations in the S protein, L54F, K77M and R88M were found to reside within the NTD domain of the S1 subunit and may have a significant effect on the receptor binding ability of the S1 subunit [33]. Two mutations, E583D and Q677H, were observed in the linker region between the S1 and S2 subunits and may influence host-protease-mediated cleavage of the spike protein during entry of SARS-CoV-2 into the cell [33]. The genomic integrity of SARS-CoV-2 principally relies on the functional efficiency of RdRp/NSP12. We observed the presence of A97V and L329I changes in the NiRaN domain, V880I in the thumb domain, and G571S in the finger domain of RdRP, which could compromise its replication fidelity and also alter its sensitivity to inhibitors such as remdesivir, ribavirin and favipiravir, which are recommended for COVID-19 treatment [34]. The S194L mutation resides in the central region of the N protein, which is essential for its oligomerization [35, 36]. We observed the co-dominance of four mutations (C241T/5’-UTR, D614G/S, F106F/NSP3 and P323L/RdRp) in all geographic regions of India. This is followed by a group of five co-dominating mutations (L37F/NSP6, T1198K/NSP3, A97V/RdRp, Y789Y/S and P13L/N), which were nearly as prevalent as the four dominant mutations in South and North India.
Traditionally, rapidly mutating positive-sense single-stranded RNA viruses undergo more transitions in their genomes than transversions [37]. Consistent with this, we observed a 3.07 times higher frequency of transitions over transversions in the SARS-CoV-2 genome. Out of the 33 different mutations, 14 were transversions, and 19 were transitions. As transversion events radically change the properties (size/charge/polarity) of the substituted amino acid, any such mutation in the coding region could affect protein function. It was not surprising to observe five transversion mutations in the S protein, because viral surface proteins often undergo a large number of mutations, leading to altered functions. This might contribute to immune evasion, as neutralizing antibodies are often generated against surface protein epitopes, some of which are important for vaccines.
A detailed analysis of the mutations showed that the C>T transitions accounted for 12 of the 34 mutations (considering RG203KR as R203K and G204R), followed in frequency by G>T (8/34), G>A (6/34), and C>A (4/34). The fewest amino acid changes occurred due to A>G (2/34), A>T (1/34), T>C (1/34), and G>C (1/34) substitutions. This is consistent with the analysis done by Ugurel et al. [38], who found C-to-T transitions and G-to-T transversions accounted for the majority of mutations in contrast to Pathan et al. [39], who found T-to-A transversions to dominate. The high frequency of C>T and G>A transitions could be mediated by APOBEC (apolipoprotein B mRNA editing catalytic polypeptide-like) enzymes, a family of cytidine deaminases that catalyze C-to-U deamination [40]. Furthermore, most of the C>T changes were identified at the second position of the codon (e.g., GCN [Ala] to GUN [Val], CCN [Pro] to CUN [Leu] and UCN [Ser] to UUN [Leu/Phe]), further underscoring the role of APOBECs, which prefer 5’-NCU-3’ sites for their action [41]. All of the synonymous mutations resulted from C>T transitions occurring at the third position of the codon. The A>G mutation (responsible for the A2-clade-specific D614G mutation) and the T>C mutation (responsible for the B-clade-specific L84S mutation) could have arisen as a result of the ADAR (adenosine deaminase acting on RNA) effect. Thus, synthetic inhibitors of APOBEC and ADAR might prove better amongst the arsenal of anti-SARS-CoV-2 drugs under trial.
Since the outbreak in Wuhan, the unceasing accumulation of genetic mutations has resulted in the formation of multiple clades and subclades originating from the prototype clade O. Coinheritance of the mutations L84S (T28144C) in ORF8 and S76S (C8782T) in NSP4 led to the emergence of clade B, which has been circulating more in the USA and less in countries of Africa and Europe. Another study had identified 1137 sequences circulating in United States that carry the P5828L mutation concurrently with L84S [42]. The world’s predominant clade, A2, emerged upon accumulation of three mutations: D614G (A23403G) in S, F106F (C3037T) in NSP3, and C241T in the 5’-UTR. In contrast to the B clade, the A2 clade was the dominant one in Europe, Africa, Asia, and Oceania but was less frequent in North and South America. After inclusion of an additional mutation, P323L (C14408T) in RdRP, into the A2 clade, a more preponderant subclade, A2a, was established. Subsequently, the G25563T (Q57H) mutation also arose in this clade. Another subclade originated from the A2a clade containing the GGG28881AAC (RG203KR) mutation. Although G25563T (Q57H) and GGG28881AAC (RG203KR) were both present in the subclades arising from the A2a clade, they were never found to co-exist. A2a subclades, with their set of co-existing mutations, were rapidly transmitted to Europe and America from their country of origin, China, establishing themselves as the dominant clades in these regions within a short time [42]. The A3 clade, which has the signature L37F (G11083T) mutation in NSP6, was found primarily in Singapore, Brunei, Thailand, Indonesia, and some parts of the Middle East, including Iraq, Iran, and Kyrgyzstan. A large number of SARS-COV-2 genomes sequenced in United Kingdom and Brazil were also found to contain the G251V mutation along with L37F, but this mutation was not significantly detected in India [18, 26, 42, 43]. The P13L mutation in N gene and the mutations S1197R and T1198K in the NSP3 gene were found to characterize the bulk of the A3 clade strains found in India.
In this study, we observed a heterogeneous distribution of SARS-CoV-2 strains of three different clades (A2a, A3 and B) in different geographic regions of India. The A2a clade (71.34%) is the major clade in all geographic regions of India, and though it contains most of the characteristic subclade mutations, such as D614G, Q57H, and RG203KR, it lacks T265I and T125M, which are present in European clades [42, 44], while having some distinct local mutations, such as S194L in N, D294D in S, S716I and A994D in NSP3, as well as some others. The A3 clade (23.29%) is India’s second most prevalent clade. It is prevalent in North and South India and is less frequent in East, West and Central India. The B clade (5.36%), the least frequent one, has been reported primarily in East and Western India. We classified the Indian isolates into 22 groups on the basis of co-existing mutations following the classification pattern of Gomez-Carballa et al. [15]. Twelve groups represented the A2a clade, with four common characteristic mutations along with various combination of novel mutations, mostly affecting the ORF3a, RdRp, S and N proteins. Eight groups aligned with the A3 clade, with a characteristic L37F mutation together with several unique mutations, mostly in nonstructural proteins (NSP2, NSP3, NSP4 and NSP12). Two groups represented B clade strains and were found to be associated with the novel mutation T749I/NSP3.
The SARS-CoV-2 genome is accumulating mutations at a very high rate. As suggested by the ‘mutation-selection balance’ and the ‘speed-fidelity trade-off’ theories [45, 46], this might be due to increased replication rate to enhance host-transmissibility at the cost of accurate replication. This might be advantageous during adaptation within a heterogeneous population where it is undergoing strong directional selection pressure due to host immunity [47]. However, other factors governing this response might be the viral genomic constellation, the presence of RNA secondary structures, the influence of host RNA editing enzymes (ADAR and APOBEC), and genetic hitchhiking [48, 49]. Not all mutations are favourable for the virus. It has been reported that when the beneficial mutations surpass the detrimental effects of the associated deleterious mutations, the deleterious mutations are subject to fixation, especially when they are encoded on the same genome segment [50, 51]. In this case, they are synonymous with respect to the non-structural proteins encoded by ORFs 1a and 1b. Although strain O was the first SARS-CoV-2 strain that was responsible for the introduction of SARS-CoV-2 into humans, it is eventually being replaced by its swarm of circulating viral quasispecies [52,53,54] in the face of host immune pressure, with the virus using a fast-replication strategy to enhance its propagation.
In conclusion, the present study highlights the rapid accumulation of various novel mutations in several proteins, principally in the S glycoprotein and the RdRp, that has led to the indigenous convergent evolution of SARS-CoV-2 circulating in different geographic regions of India. Presently, vaccine development and RdRp-inhibitor-based therapies are being targeted to control the global pandemic. However, for the development of successful therapeutics, it is imperative to monitor mutations in the targeted genes. This study has provided much-needed information regarding novel mutations in S, RdRp, and several other non-structural proteins that could pave the way for vaccine formulation and for designing antiviral drugs targeting specific viral proteins.
References
Sackman AM, McGee LW, Morrison AJ, Pierce J, Anisman J, Hamilton H et al (2017) Mutation-driven parallel evolution during viral adaptation. Mol Biol Evol 34(12):3243–3253
Barr JN, Fearns R (2016) Genetic instability of RNA viruses. In: Genome stability. Academic Press, pp 21–35
Burch CL, Chao L (1999) Evolution by small steps and rugged landscapes in the RNA virus ϕ6. Genetics 151(3):921–927
Sanjuán R, Nebot MR, Chirico N, Mansky LM, Belshaw R (2010) Viral mutation rates. J Virol 84(19):9733–9748
Koelle K, Rasmussen DA (2015) The effects of a deleterious mutation load on patterns of influenza A/H3N2’s antigenic evolution in humans. Elife. 15(4):e07361
Presti AL, Rezza G, Stefanelli P (2020) Selective pressure on SARS-CoV-2 protein coding genes and glycosylation site prediction. Heliyon. 21:e05001
Liu W, Zhang Q, Chen J, Xiang R, Song H, Shu S, Chen L, Liang L, Zhou J, You L, Wu P (2020) Detection of Covid-19 in children in early January 2020 in Wuhan, China. N Engl J Med 382(14):1370–1371
Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, Wang W, Song H, Huang B, Zhu N, Bi Y (2020) Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395(10224):565–574
Mackenzie JS, Smith DW (2020) COVID-19: a novel zoonotic disease caused by a coronavirus from China: what we know and what we don’t. Microbiol Aust 41(1):45–50
Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL, Chen HD (2020) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579(7798):270–273
Lam TT, Jia N, Zhang YW, Shum MH, Jiang JF, Zhu HC, Tong YG, Shi YX, Ni XB, Liao YS, Li WJ (2020) Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature 26:1–4
Zhang T, Wu Q, Zhang Z (2020) Probable pangolin origin of SARS-CoV-2 associated with the COVID-19 outbreak. Curr Biol 30(7):1346–1351
Drake JW, Holland JJ (1999) Mutation rates among RNA viruses. Proc Natl Acad Sci 96(24):13910–13913
Tang X, Wu C, Li X, Song Y, Yao X, Wu X, Duan Y, Zhang H, Wang Y, Qian Z, Cui J (2020) On the origin and continuing evolution of SARS-CoV-2. Natl Sci Rev 7(6):1012–1023
Gomez-Carballa A, Bello X, Pardo-Seco J, Martinon-Torres F, Salas A (2020) Mapping genome variation of SARS-CoV-2 worldwide highlights the impact of COVID-19 super-spreaders. Genome Res 30(10):1434–1448
WHO Coronavirus Disease (COVID-19) database. 30th August, 2020. https://www.covid19.who.int/
Kumar M, Taki K, Gahlot R, Sharma A, Dhangar K (2020) A chronicle of SARS-CoV-2: part-I-epidemiology, diagnosis, prognosis, transmission and treatment. Sci Total Environ 15:139278
Maitra A, Sarkar MC, Raheja H, Biswas NK, Chakraborti S, Singh AK (2020) Mutations in SARS-CoV-2 viral RNA identified in Eastern India: possible implications for the ongoing outbreak in India and impact on viral structure and host susceptibility. J Biosci 45(1):76
Banu S, Jolly B, Mukherjee P, Singh P, Khan S, Zaveri L, Shambhavi S, Gaur N, Reddy S, Kaveri K, Srinivasan S, Gopal DR, Siva AB, Thangaraj K, Tallapaka KB, Mishra RK, Scaria V, Sowpati DT (2020) A Distinct Phylogenetic Cluster of Indian Severe Acute Respiratory Syndrome Coronavirus 2 Isolates. In Open Forum Infect Dis 7(11):ofaa434
Du X, Wang Z, Wu A, Song L, Cao Y, Hang H et al (2008) Networks of genomic co-occurrence capture characteristics of human influenza A (H3N2) evolution. Genome Res 18(1):178–187
Jensen JD, Lynch M (2020) Considering mutational meltdown as a potential SARS-CoV-2 treatment strategy. Heredity 124(5):619–620
Wu D, Wu T, Liu Q, Yang Z (2020) The SARS-CoV-2 outbreak: what we know. Int J Infect Dis 94:44–48
Shu Y, McCauley J (2017) GISAID: global initiative on sharing all influenza data—from vision to reality. Eurosurveillance 22(13):30494
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 60. Mol Biol Evol 30(12):2725–2729
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32(5):1792–1797
Mercatelli D, Giorgi FM (2020) Geographic and genomic distribution of SARS-CoV-2 mutations. Front Microbiol 11:1800
Guan Q, Sadykov M, Mfarrej S, Hala S, Naeem R, Nugmanova R, Al-Omari A, Salih S, Al Mutair A, Carr MJ, Hall WW (2020) A genetic barcode of SARS-CoV-2 for monitoring global distribution of different clades during the COVID-19 pandemic. Int J Infect Dis 100:216–223
Hassan SS, Choudhury PP, Roy B, Jana SS (2020) Missense mutations in SARS-CoV2 genomes from Indian patients. Genomics 112(6):4622–4627. https://doi.org/10.1016/j.ygeno.2020.08.021
Phan T (2020) Genetic diversity and evolution of SARS-CoV-2. Infect Genet Evol 1(81):104260
Becerra‐Flores M, Cardozo T (2020) SARS‐CoV‐2 viral spike G614 mutation exhibits higher case fatality rate. Int J Clin Pract 74:e13525
Korber B, Fischer WM, Gnanakaran S, Yoon H, Theiler J, Abfalterer W, Hengartner N, Giorgi EE, Bhattacharya T, Foley B, Hastie KM (2020) Tracking changes in SARS-CoV-2 Spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell 182(4):812–827
Hu J, He CL, Gao Q, Zhang GJ, Cao XX, Long QX et al (2020) The D614G mutation of SARS-CoV-2 spike protein enhances viral infectivity. BioRxiv. https://doi.org/10.1101/2020.06.20.161323
Wang Q, Zhang Y, Wu L, Niu S, Song C, Zhang Z et al (2020) Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 181:1–11
Kirchdoerfer RN, Ward AB (2019) Structure of the SARS-CoV nsp12 polymerase bound to nsp7 and nsp8 co-factors. Nat Commun 10(1):1–9
Yu IM, Oldham ML, Zhang J, Chen J (2006) Crystal structure of the severe acute respiratory syndrome (SARS) coronavirus nucleocapsid protein dimerization domain reveals evolutionary linkage between corona-and arteriviridae. J Biol Chem 281(25):17134–17139
Zhao P, Cao J, Zhao LJ, Qin ZL, Ke JS, Pan W et al (2005) Immune responses against SARS-coronavirus nucleocapsid protein induced by DNA vaccine. Virology 331(1):128–135
Sanjuán R, Domingo-Calap P (2016) Mechanisms of viral mutation. Cell Mol Life Sci 73(23):4433–4448
Uğurel O, Ata O, Balik D (2020) An updated analysis of variations in SARS-CoV-2 genome. Turk J Biol 44(SI-1):157–167
Pathan RK, Biswas M, Khandaker MU (2020) Time series prediction of COVID-19 by mutation rate analysis using recurrent neural network-based LSTM model. Chaos Solitons Fract 13:110018
Di Giorgio S, Martignano F, Torcia MG, Mattiuz G, Conticello SG et al (2020) Evidence for host-dependent RNA editing in the transcriptome of SARS-CoV-2. Sci Adv 6(15):eabb5813
Rosenberg BR, Hamilton CE, Mwangi MM, Dewell S, Papavasiliou FN et al (2011) Transcriptome-wide sequencing reveals numerous APOBEC1 mRNA-editing targets in transcript 3′ UTRs. Nat Struct Mol Biol 18(2):230
Koyama T, Platt D, Parida L (2020) Variant analysis of SARS-CoV-2 genomes. Bull World Health Organ 98(7):495
Gonzalez-Reiche AS, Hernandez MM, Sullivan MJ, Ciferri B, Alshammary H, Obla A et al (2020) Introductions and early spread of SARS-CoV-2 in the New York City area. Science 369(6501):297–301
Jones LR, Manrique JM (2020) Quantitative phylogenomic evidence reveals a spatially structured SARS-CoV-2 diversity. Virology 1(550):70–77
Regoes RR, Hamblin S, Tanaka MM (2013) Viral mutation rates: modelling the roles of within-host viral. Proc Biol Sci 280(1750):20122047
Fitzsimmons WJ, Woods RJ, McCrone JT, Woodman A, Arnold JJ, Yennawar M et al (2018) A speed–fidelity trade-off determines the mutation rate and virulence of an RNA virus. PLoS Biol 16(6):e2006459
Duffy S (2018) Why are RNA virus mutation rates so damn high? PLoS Biol 16(8):e3000003
Sanjuán R, Thoulouze MI (2019) Why viruses sometimes disperse in groups. Virus Evol 5(1):vez014
Combe M, Sanjuan R et al (2014) Variation in RNA virus mutation rates across host cells. PLoS Pathog 10(1):e1003855
Zanini F, Neher RA (2013) Quantifying selection against synonymous mutations in HIV-1 env evolution. J Virol 87(21):11843–11850
Stern A, Bianco S, TeYeh M, Wright C, Butcher K, Tang C et al (2014) Costs and benefits of mutational robustness in RNA viruses. Cell Rep 8(4):1026–1036
Silander OK, Tenaillon O, Chao L (2007) Understanding the evolutionary fate of finite populations: the dynamics of mutational effects. PLoS Biol 5(4):e94
Peck KM, Lauring AS (2018) Complexities of viral mutation rates. J Virol 92(14):e01031-e1117
Domingo E, Perales C (2019) Viral quasispecies. PLoS Genet 15(10):e1008271
Acknowledgements
This study was supported by the Indian Council of Medical Research, India. RS is supported by a senior research fellowship from UGC. The authors acknowledge the hard work of scientists and laboratory staffs in all of the COVID-19 testing laboratories and next-gen sequencing labs across India.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sector.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that no conflict of interest exists.
Additional information
Handling Editor: Zhenhai Chen.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary Fig. 1
Pie chart representation of strains from different geographic regions taken into consideration for mutation analysis. The states belonging to each such region and the numbers of strains analyzed are indicated (TIF 2507 KB)
Rights and permissions
About this article

Cite this article
Sarkar, R., Mitra, S., Chandra, P. et al. Comprehensive analysis of genomic diversity of SARS-CoV-2 in different geographic regions of India: an endeavour to classify Indian SARS-CoV-2 strains on the basis of co-existing mutations. Arch Virol 166, 801–812 (2021). https://doi.org/10.1007/s00705-020-04911-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-020-04911-0