
Abstract
Microglial activation is an early phenomenon in Alzheimer’s disease (AD) that may occur prior to and independently of amyloid-β (Aβ) aggregation. Recent studies in transgenic animal models suggest that the apolipoprotein E ε4 (APOEε4) allele may be a culprit of early microglial activation in AD. However, it is unclear whether the APOEε4 genotype is associated with microglial reactivity in the living human brain. Here, we tested whether APOEε4 carriership is associated with microglial activation in individuals across the aging and AD spectrum. We studied 118 individuals who had positron emission tomography (PET) for Aβ ([18F]AZD4694), tau ([18F]MK6240), and microglial activation ([11C]PBR28), as well as clinical, genetic, and magnetic resonance imaging data. We found that APOEε4 carriership was associated with increased microglial activation mainly in early Braak-staging regions within the medial temporal cortex, and this effect of APOEε4 was independent of Aβ and tau deposition. Furthermore, microglial activation mediated the Aβ-independent effects of APOEε4 on downstream tau accumulation, neurodegeneration, and clinical impairment. Interestingly, the physiological distribution of APOE mRNA expression, obtained from the Allen Human Atlas, predicted the patterns of APOEε4-related microglial activation in our population, suggesting that the deleterious effects of APOEε4 occur at the level of gene expression. These results support a model in which the APOEε4 has Aβ-independent effects on AD pathogenesis by activating microglia in brain regions associated with early tau deposition. Our findings provide a rationale for the development of novel AD therapies targeting the interplay between ApoE and neuroinflammation.
Introduction
Alzheimer’s disease (AD) is a multifactorial disorder neuropathologically characterized by amyloid-β (Aβ) plaques and tau neurofibrillary tangles (1, 2). Among the multiple pathogenic processes involved in AD etiology, neuroinflammation - commonly associated with microglial reactivity - has been increasingly recognized (3, 4). Microglial activation plays a key role in the accumulation of AD hallmark proteinopathies, rather than being merely an epiphenomenon of their deposition (3, 4). Specifically, recent observations from animal and human studies suggest that microglial activation precedes and may drive tau spread over the neocortex following a Braak stage-like pattern (5-7), from the medial temporal to association and primary sensory structures (8-11). Such microglial activation is synaptotoxic, affects brain connectivity, and predicts clinical decline (12, 13). Aβ pathology can trigger microglial activation in AD (14-16), but Aβ plaques and activated microglia only partially overlap topographically in the human brain (17-19), and microglial activation may occur prior to demonstrable Aβ deposition (3).
The apolipoprotein E ε4 (APOEε4) allele is a major genetic risk factor for sporadic AD (20-23). The link between APOEε4 and Aβ deposition is an important factor leading to AD progression (24). However, recent animal studies suggest that the APOEε4 genotype may also contribute to AD pathogenesis through Aβ-independent pathways by potentiating brain inflammation, tau accumulation, and neurodegeneration (25, 26). Although robust experimental evidence indicates that the APOE genotype modulates microglial response in AD (25, 27-30), it remains to be elucidated whether APOEε4 carriership is associated with microglial activation in the AD human brain. The characterization of this association in living individuals is critical to confirm the Aβ-independent detrimental effects of APOEε4 on microglia homeostasis and to support the development of novel therapeutic strategies.
Using complementary positron emission tomography (PET) radiotracers for the topographical quantification of microglial activation, Aβ, and tau accumulation across the brain, we investigated the association of APOEε4 carriership, microglial activation, Aβ, and tau in a cohort of individuals across the aging and AD continuum. We hypothesized that APOEε4 is associated with microglial activation independently of AD hallmark proteinopathies. We then tested whether microglial activation mediates the effects of APOEε4 on tau accumulation, neurodegeneration, and clinical impairment. Postmortem data from the Allen Human Brain Atlas was used to test the link between regional levels of brain APOE gene expression and the distribution of microglial activation as a function of APOEε4 carriership.
Results
Participants
We screened 606 people for the rs6971 polymorphism in the translocator protein (TSPO) gene. Out of the 314 high-affinity binders, we studied 118 individuals that were across the aging and AD spectrum (79 cognitively unimpaired [CU], 23 with mild cognitive impairment [MCI], and 16 with AD dementia) with [18F]AZD4694 Aβ PET, [18F]MK6240 tau PET, [11C]PBR28 microglial activation PET, and magnetic resonance imaging (MRI), as well as APOE genotyping (Fig. S1). Demographic characteristics of the population are reported in Table 1. Information regarding the prevalence of APOE genotypes in our sample is described in Table S1.
APOEε4 associates with microglial activation in the medial temporal cortex
To test the association between APOEε4 status and [11C]PBR28 microglial activation PET, we conducted linear regression analyses adjusting for age, sex, and clinical diagnosis. Voxel-wise regression analysis showed that APOEε4 carriership was associated with higher [11C]PBR28 uptake mainly in medial temporal structures (transentorhinal, entorhinal, and hippocampal cortices), which are regions corresponding to early Braak stages (Fig. 1A, B). Regarding the spatial extent of the voxel-wise results, the association between APOEε4 carriership and [11C]PBR28 (SUVR) was predominantly observed in areas corresponding to Braak I (affecting 94.8% of this region), followed by Braak II (47.6%), Braak III (16.7%), Braak IV (13.1%), Braak V (2.5%), and Braak VI regions (1.0%; Fig. 1C). Similarly, in terms of the magnitude of the associations (β estimate), the relationship between APOEε4 and [11C]PBR28 uptake was progressively weaker from Braak I to VI, surviving Bonferroni correction for multiple comparisons only in Braak I and II regions (Braak I: β = 0.088, 95% confidence interval [CI] 0.044 to 0.133, P < 0.001; Braak II: β = 0.066, 95% CI 0.020 to 0.111, P = 0.005; Fig. 1D). Sensitivity analysis excluding individuals bearing the ε2 allele of the APOE gene showed similar findings (Fig. S2).

(A) T-map shows the result of voxel-wise linear regression testing the association of APOEε4 carriage status (noncarrier or carrier) with [11C]PBR28 SUVR accounting for age, sex, and clinical diagnosis (CU, MCI, or AD). Results survived random field theory correction for multiple comparisons at P < 0.05. (B) Bars show the mean and standard deviation of [11C]PBR28 SUVR in APOEε4 noncarriers and carriers. Imaging biomarker values were extracted from the peak T-value cluster of the voxel-wise analysis (T-value ≥ 4.7). P-value indicates the result of regression analysis accounting for age, sex, and clinical diagnosis. (C) Bars represent the spatial extent of the APOEε4-related microglia activation across Braak regions. Values were calculated by determining the percentage of voxels per Braak region having an association (T-value > 2) between APOEε4 carriership and [11C]PBR28 in the voxel-wise analysis. (D) β estimates with 95% CI represent the strength of the regional association between APOEε4 status and [11C]PBR28 SUVR across Braak regions from ROI-based linear regressions. Models were adjusted for age, sex, and clinical diagnosis. Estimates that survived Bonferroni correction at P < 0.05 are indicated with a double asterisk.
APOEε4 associates with microglial activation independently of Aβ and tau biomarkers
We investigated whether the association between APOEε4 carriership and [11C]PBR28 uptake was independent of AD hallmark proteinopathies by correcting the regression models for global [18F]AZD4694 Aβ PET and local [18F]MK6240 tau PET, as well as age, sex, and clinical diagnosis. APOEε4 carriership was associated with higher [11C]PBR28 uptake in Braak I-II regions independently of global Aβ and local tau PET (β = 0.055, 95% CI 0.010 to 0.100, P = 0.018; Table 2). In a subgroup of 51 participants (31 CU, 12 with MCI, and 8 with AD dementia), we conducted sensitivity analyses adjusting for cerebrospinal fluid (CSF) Aβ1-42 and p-tau181 instead of [18F]AZD4694 Aβ PET and [18F]MK6240 tau PET, respectively. Demographics for the subgroup of participants are presented in Table S2. Similarly, we found that APOEε4 carriership was significantly associated with higher [11C]PBR28 SUVR in Braak I-II regions independently of CSF Aβ1-42 and p-tau181 (β = 0.073, 95% CI 0.005 to 0.140, P = 0.035; Table S3), which reinforces the cross-modality imaging results. Exploratory analyses conducted across the six Braak regions supported that the Aβ- and tau-independent association of APOEε4 carriership with [11C]PBR28 uptake was mainly confined to early Braak regions, either assessing AD hallmark proteins with imaging ([18F]AZD4694 Aβ PET and [18F]MK6240 tau PET; Table S4) or fluid biomarkers (CSF Aβ1-42 and p-tau181; Table S5).
APOE gene expression resembles APOEε4-related microglial activation patterns
We studied the topographical distribution of APOE messenger RNA (mRNA) in the postmortem brain of six CU individuals from the Allen Human Brain Atlas. We observed different APOE gene expression levels across Braak regions, which were progressively lower from Braak I to VI (Fig. 2A), suggesting that the cerebral levels of APOE gene expression partially follow Braak-like stages. Additionally, linear regressions demonstrated that the regional patterns of Allen APOE mRNA expression across Braak regions predicted the topography and magnitude of APOEε4 effects on [11C]PBR28 uptake observed in our population (Fig. 2B, C).

(A) Brain map of the topographical distribution of APOE mRNA expression in 6 CU individuals obtained from the Allen Human Brain Atlas (left). Average intensity values of APOE mRNA expression in each Braak region (right). (B) Regression analysis testing whether Allen APOE gene expression patterns predict the percentage of the area showing APOEε4-related [11C]PBR28 SUVR increase across Braak regions in our population. (C) Regression analysis testing whether the Allen brain APOE gene expression patterns predict the magnitude/strength of the association between APOEε4 and [11C]PBR28 uptake across Braak regions in our population.
Microglial activation mediates the Aβ-independent effects of APOEε4 on downstream AD markers
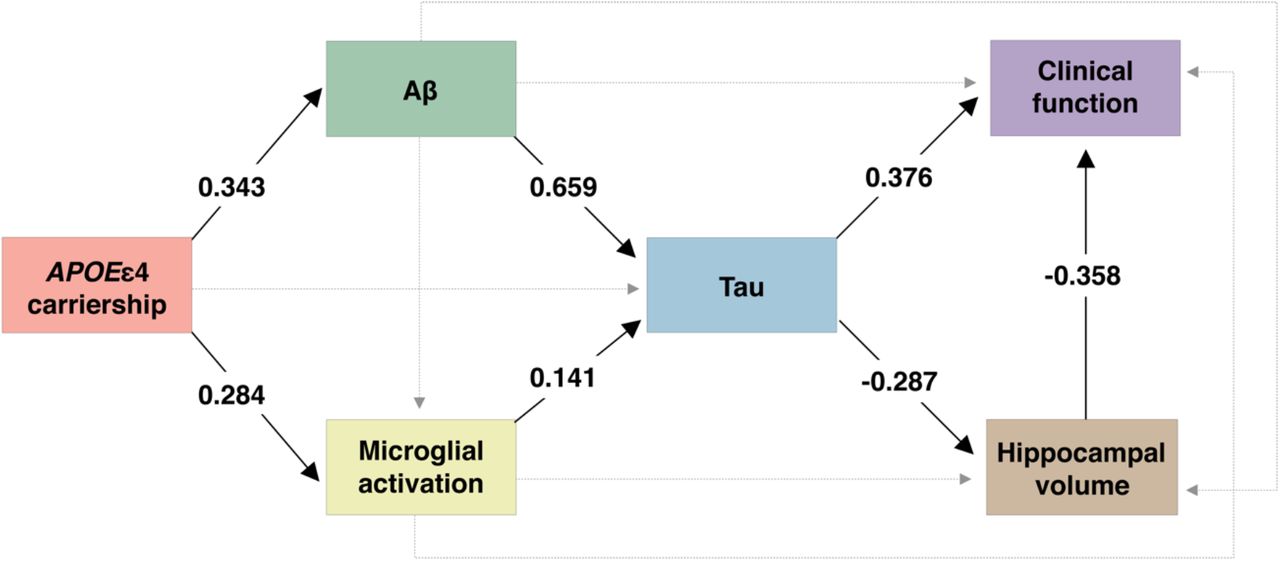
We next used structural equation modeling to investigate the associations between APOEε4, microglial activation, Aβ, tau, hippocampal volume, and clinical function (as measured with the clinical dementia rating scale sum of boxes [CDR-SB] score). We found that an increase in [11C]PBR28 microglial activation uptake partially mediated the effects of APOEε4 carriership on higher tau PET uptake in Braak I-II regions independently of Aβ PET burden. The model also showed the well-reported effects of APOEε4 on higher tau PET uptake through higher Aβ PET load, which were independent of microglial activation. Notably, both Aβ-independent and -dependent pathways leading to medial temporal tau pathology were further associated with lower hippocampal volume and, ultimately, higher severity of clinical impairment (Fig. 3). This model fit the data well (n = 118, X2 = 7.141, degrees of freedom = 5, P = 0.210, root mean squared error of approximation [RMSEA] = 0.060, standardized root mean square residual [SRMR] = 0.023, comparative fit index [CFI] = 0.991). Complete model coefficients and associated statistics are described in Table S6.

{kind=link}
{kind=link}
{kind=link}
The values presented in the figure are structural equation model β estimates testing the associations between APOEε4 status, microglial activation PET, Aβ PET, tau PET, hippocampal volume, and clinical function. Given that the β estimates presented in the figure are standardized, the effects can be directly compared. Solid lines represent significant associations, whereas dashed lines represent non-significant effects. All associations were adjusted for age and sex. Associations involving hippocampal volume and clinical function were also adjusted for years of education. Aβ pathology was measured with global [18F]AZD4694 SUVR, microglial activation with Braak I-II [11C]PBR28 SUVR, and tau pathology with Braak I-II [18F]MK6240 SUVR. Clinical function was assessed with the CDR-SB score.
Discussion
In the present study, we observed that APOEε4 carriership was associated with microglial activation in early Braak-stage regions independently of AD hallmark proteinopathies. We also found that the brain distribution of APOE gene expression predicted the pattern of APOEε4-related microglial activation observed in our study population. Lastly, we demonstrated that microglial activation partially mediated the APOEε4 effects on regional tau accumulation through an Aβ-independent pathway, which was further associated with neurodegeneration and clinical impairment. Taken together, our findings support the hypothesis that APOEε4 contributes to the early progression of AD via increased neuroinflammation.
APOEε4 carriership was associated with higher levels of microglial activation in living humans across the aging and AD continuum. Several recent investigations in animal models of AD support our findings. For example, the APOE genotype modulates microglial response in AD, with APOEε4 being associated with multiple microglial-related detrimental downstream effects (e.g., protein aggregation, neurodegeneration, and dysfunctional immunometabolic response) (25, 27-30). Further experiments showed that the APOEε4 genotype associates with changes in the transcriptional profile of microglia from a homeostatic state to a disease-associated state across AD progression (31, 32) and that the activation of microglial-related proteins (e.g., triggering receptor expressed on myeloid cells 2 (TREM2) (33-36)) is directly associated with ApoE signaling (37). This evidence raises the possibility that the mechanisms explaining the link between ApoE and microglial activation occur at the transcriptional level by the expression of microglia-specific genes such as TREM2. Altogether, the aforementioned findings suggest that the APOEε4 genotype is associated with a disruption in microglia homeostasis in AD, promoting disease-associated microglia that plays a role in the development of the disease.
Our results showed an effect of APOEε4 on medial temporal microglial activation leading to AD progression that is partially independent of Aβ and tau. Previous observations indicate that microglial activation may precede and drive tau propagation (5-7, 38). Moreover, an investigation using the [11C]PK11195 PET tracer found that microglial activation in the temporal lobe is associated with longitudinal cognitive decline more strongly than the [18F]AV1451 tau PET tracer binding in patients presenting AD pathophysiology (12). We complemented these reports by demonstrating an Aβ-independent effect of APOEε4 on early microglial activation in medial temporal structures, which in turn mediate tau accumulation and AD progression. These results resonate with recent CSF biomarker evidence of neuroinflammation in adult APOEε4 carriers who have not developed Aβ pathology yet (39). Our findings are also in line with animal model studies showing that the APOE genotype affects tau pathology as well as tau-mediated neurodegeneration independently of Aβ, with the APOEε4 isoform having detrimental effects on both outcomes (25, 38, 40-42). In humans, APOEε4 carriership has been associated with medial temporal atrophy (43-46). Additionally, a recent study revealed that APOEε4 carriership is associated with tau PET uptake in the medial temporal lobe independently of Aβ, raising discussions about the importance of elucidating the biological underpinnings of this association (47). We built on these previous investigations suggesting that microglial activation is the mediator of the Aβ-independent effects of APOEε4 on medial temporal tau deposition and brain atrophy, leading to dementia. Together, these results suggest that disease-modifying therapies targeting the interplay between ApoE and microglial activation have the potential to slow downstream AD progression.
Interestingly, we observed that cerebral APOE mRNA expression was more prominent in medial temporal structures and its levels hierarchically followed the Braak staging scheme. Even though the Allen Human Brain Atlas is derived from younger adults without dementia, the APOE gene expression pattern was able to predict the topography and magnitude of the APOEε4-related [11C]PBR28 uptake increase in our cohort. It is well-established that tau neurofibrillary tangles follow a stereotypical pattern of progression known as Braak stages, with tau tangles deposition starting in the medial temporal cortex (8-11). Microglial activation precedes and drives tau spread from the medial temporal lobe to the neocortex in a Braak-like pattern in AD models (5-7), although the mechanism associated with triggering microglial activation in medial temporal structures was not fully understood. Here, we first showed that APOEε4 plays a role in triggering microglia activation in early Braak regions, and second that a Braak-like pattern of APOE gene expression could shed light on the entire hierarchical progression of tau across these stages. These results indicate that microglia-mediated tau propagation in AD might be explained at least partially by the cerebral expression levels of ApoE4.
Methodological strengths of the present work include the assessment of a well-characterized cohort with multiple PET radiotracers acquired on the same scanner, which allows high-quality topographical characterization of microglial activation, Aβ deposition, and tau accumulation using the best currently available technologies. Moreover, we screened a large sample of 606 individuals for the rs6971 polymorphism in the TSPO gene, and, consequently, we were able to include only high-affinity binders for the [11C]PBR28 radiotracer, which increases the signal-to-noise ratio of the tracer and the reliability of our results. Methodological limitations need to be acknowledged and considered to interpret our results. The [11C]PBR28 radiotracer binds to the TSPO, which is a protein mainly expressed in the mitochondrial outer membrane of activated microglia (3). Thus, [11C]PBR28 PET is considered an imaging biomarker of a general cerebral microglial activation state (7, 48, 49). However, it is recognized that microglia may acquire diverse phenotypes across disease progression (3), and this heterogeneity cannot be captured using the available human brain imaging technologies. Furthermore, it is possible that other cell types (e.g., astrocytes) also play a minor role in the TSPO PET signal (3, 50-53). Because CSF measures have been suggested to detect Aβ and tau accumulation earlier than PET (48, 54, 55), the fact we observed similar results using imaging ([18F]AZD4694 and [18F]MK6240) and fluid (CSF Aβ1-42 and p-tau181) biomarkers support the robustness of our findings. However, we cannot exclude a possible contribution of pre-fibrillary Aβ and tau aggregates to our results. Future studies using multiple longitudinal measures are needed to better evaluate the sequential relationship between neuroimaging biomarkers. Lastly, individuals included in our investigation were volunteers motivated to participate in a study about AD, which can be a source of self-selection bias.
In conclusion, our results support the existence of an Aβ-independent effect of APOEε4 on AD progression through microglial activation, leading to tau accumulation, neurodegeneration, and eventually clinical impairment.
Materials and methods
Experimental design
The main objective of the present study was to test the association between APOEε4 carriership and brain levels of microglial activation. We hypothesized that APOEε4 is associated with microglial activation in early Braak regions independently of Aβ and tau pathologies. Furthermore, we aimed to assess whether microglial activation mediates the effects of APOEε4 on downstream AD progression. Participants from the community or outpatients at the McGill University Research Centre for Studies in Aging were enrolled in the Translational Biomarkers in Aging and Dementia (TRIAD) study (https://triad.tnl-mcgill.com). Participants were required to have adequate visual and auditory capacities for neuropsychological assessment, as well as the ability to speak English or French. Additionally, individuals were not enrolled if they had active substance abuse, major surgery, recent head trauma, neuroimaging contraindication, simultaneously being enrolled in other studies, and untreated neurological, psychiatric, or systemic conditions. This study was approved by the Douglas Mental Health University Institute Research Ethics Board and the Montreal Neurological Institute PET working committee, and all participants provided written informed consent.
Participants
Out of the 606 individuals screened for the rs6971polymorphism in the TSPO gene, we studied 118 individuals with high-affinity binding aged 52 to 87 years (79 CU, 23 with MCI, and 16 with AD dementia). At the same time point, all participants had PET for Aβ ([18F]AZD4694), tau tangles ([18F]MK6240), and microglial activation ([11C]PBR28), as well as MRI and APOE genotyping. Two individuals that had [11C]PBR28 or [18F]MK6240 SUVR values three standard deviations (SD) above the mean of the population were considered outliers as defined a priori and excluded from the analyses. A flowchart describing the selection of study participants is reported in Fig. S1. Participants underwent detailed neuropsychological assessments, including Mini-Mental State Examination (MMSE) and Clinical Dementia Rating (CDR). CU subjects had no objective cognitive impairment and a global CDR score of 0. MCI patients had subjective and objective cognitive impairment, preserved activities of daily living, and a global CDR score of 0.5 (56). Mild-to-moderate AD dementia patients had a global CDR score between 0.5 and 2 and met the National Institute on Aging and the Alzheimer’s Association (NIA-AA) criteria for probable AD (57). We analyzed all the individuals with complete data, and no power analysis was performed before the study. It is worth noting that the sample size of the present work is similar to the largest previous TSPO PET studies across the AD spectrum (7, 58-60).
Genetic data
[11C]PBR28 binding affinity is influenced by a common polymorphism (rs6971) in the TSPO gene (https://www.ncbi.nlm.nih.gov/snp/rs6971). In order to increase the reliability of our results, we genotyped 606 participants for the rs6971 polymorphism before imaging, and we only included high-affinity binders (7). Noteworthy, this polymorphism is a methodological caveat and does not affect TSPO levels, glial activity, or AD pathological changes (53). Moreover, subjects were genotyped for the APOE gene using the polymerase chain reaction amplification technique, followed by restriction enzyme digestion, standard gel resolution, and visualization processes (61).
Brain imaging
T1-weighted MRIs were acquired at the Montreal Neurological Institute (MNI) using a 3T Siemens Magnetom. We used the magnetization prepared rapid acquisition gradient echo (MPRAGE) MRI (TR: 2300 ms, TE: 2.96ms) sequence to obtain high-resolution structural images of the whole brain (9° flip angle, coronal orientation perpendicular to the double spin echo sequence, 1×1 mm2 in-plane resolution of 1 mm slab thickness) (62). Aβ PET with [18F]AZD4694 (40–70 min post-injection), tau PET with [18F]MK-6240 (90–110 min post-injection), and microglial activation TSPO PET with [11C]PBR28 (60–90 min post-injection) were acquired at the MNI using a Siemens High Resolution Research Tomograph. PET scans were reconstructed using the ordered subset expectation maximization (OSEM) algorithm on a four-dimensional (4D) volume with three frames (3 × 600 seconds) for [18F]AZD4694 PET (63), four frames (4 × 300 seconds) for [18F]MK-6240 PET (63), and six frames (6 × 300 s) for [11C]PBR28 PET (7). Then, attenuation correction was performed using a 6-minute transmission scan with a rotating 137Cs point source. Furthermore, PET images were corrected for motion, dead time, decay, and scattered and random coincidences. Following an in-house pipeline, T1-weighted MRIs were corrected for non-uniformity and field distortion. Subsequently, linear co-registration and non-linear spatial normalization for the ADNI template were performed through linear and non-linear transformation in two main steps: (i) PET registration to the correspondent T1-weighted MRI and (ii) T1-weighted MRI registration to the ADNI reference space. PET images were spatially smoothed to achieve a final resolution of 8 mm full width at half-maximum. SUVRs were calculated using the whole cerebellum gray matter for [18F]AZD4694 Aβ PET (64) and [11C]PBR28 microglial activation PET (7), and the inferior cerebellum gray matter for [18F]MK-6240 tau PET (65). The Desikan-Killiany-Tourville atlas was used to determine the regions of interest (ROIs) (66). A global Aβ PET SUVR was estimated from the precuneus, prefrontal, orbitofrontal, parietal, temporal, and cingulate cortices (67). Based on postmortem observations (8, 9) and PET studies (65, 68), PET Braak-like stages were calculated from brain regions corresponding to the Braak stages of tau neurofibrillary tangle accumulation: Braak I (transentorhinal), Braak II (entorhinal and hippocampus), Braak III (amygdala, parahippocampal gyrus, fusiform gyrus, lingual gyrus), Braak IV (insula, inferior temporal, lateral temporal, posterior cingulate, and inferior parietal), Braak V (orbitofrontal, superior temporal, inferior frontal, cuneus, anterior cingulate, supramarginal gyrus, lateral occipital, precuneus, superior parietal, superior frontal, rostro medial frontal), and Braak VI (paracentral, postcentral, precentral, and pericalcarine) (65, 69). A representation of the Braak-like regions used in our analysis is shown in Fig. S3. Hippocampal volume was adjusted for total intracranial volume using MRI data from CU subjects (70).
The Allen Human Brain Atlas (http://www.brain-map.org) (71) was used to obtain information regarding APOE gene expression in the brain. In brief, microarray was used to calculate mRNA expression intensity values on 3702 samples from 6 healthy human postmortem brains (4 males, mean age = 42.5 (13.4) years, postmortem delay = 20.6 (7) hours). The APOE mRNA brain expression maps were derived from a Gaussian process (72) and downloaded from www.meduniwien.ac.at/neuroimaging/mRNA.html.
CSF measurements
A subgroup of 51 individuals had CSF Aβ1-42 and p-tau181 quantified using the LUMIPULSE G1200 instrument (Fujirebio) at the Clinical Neurochemistry Laboratory, University of Gothenburg, Mölndal, Sweden (73).
Statistical analysis
Analyses were performed in the R software (version 4.0.2, http://www.r-project.org/). Neuroimaging analyses were conducted using RMINC (74), an imaging package that allows the integration of voxel-based statistics into the R statistical environment. Voxel-wise and ROI-based linear regressions tested the association between APOEε4 status (noncarrier or carrier) and microglial activation indexed by the [11C]PBR28 SUVR adjusting for age, sex, and clinical diagnosis (CU, MCI, or AD). To further investigate whether this association was independent of AD hallmark proteins, ROI-based models were also adjusted for Aβ ([18F]AZD4694 PET or CSF Aβ1-42) and tau ([18F]MK-6240 PET or CSF p-tau181) biomarkers. Multiple comparison correction at P < 0.05 was performed using random field theory for voxel-wise analysis and Bonferroni method for ROI-based analysis when appropriate. We calculated the percentage of voxels in each Braak region showing an association (T-value > 2) between APOEε4 carriership and [11C]PBR28 SUVR in the aforementioned voxel-wise analysis. Regression analysis tested whether the APOE mRNA expression intensity predicted the topography and magnitude of APOEε4-related [11C]PBR28 SUVR increase across Braak regions. Structural equation modeling, R package “lavaan” (75), was used to test the associations between APOEε4 status, microglial activation, Aβ, tau, hippocampal volume, and clinical function (as measured with the CDR-SB score). All the associations in the model were adjusted for age and sex; associations involving hippocampal volume and clinical deterioration were further adjusted for years of education. Structural equation model was judged as having a good fit as follows: CFI > 0.97 (acceptable: 0.95 - 0.97); RMSEA < 0.05 (acceptable 0.05 - 0.08); SRMR < 0.05 (acceptable: 0.05 - 0.10) (76, 77). Statistical significance of parameters estimates from the structural equation model was tested using bootstrapping with 1000 permutations. For all analyses, two-tailed P-values < 0.05 were considered statistically significant.
Data Availability
The data used in the present work is not publicly available as the information could compromise the participants′ privacy. Therefore, the data from the TRIAD study will be made available from the senior authors upon reasonable request, and such arrangements are subject to standard data-sharing agreements.
List of Supplementary Materials
Fig. S1. Flowchart of included participants.
Fig. S2. Sensitivity analysis excluding APOEε2 carriers.
Fig. S3. Braak regions mask used in the analyses.
Table S1. Prevalence of APOE genotypes.
Table S2. Demographics of the subsample with available CSF biomarkers.
Table S3. Sensitivity analysis testing the association of APOEε4 carriership with microglial activation adjusting for CSF Aβ1-42 and p-tau181.
Table S4. Associations between microglial activation and APOEε4 carriership across all Braak regions adjusting for global [18F]AZD4694 Aβ PET and local [18F]MK6240 tau PET.
Table S5. Associations between microglial activation and APOEε4 carriership across all Braak regions adjusting for CSF Aβ1-42 and p-tau181.
Table S6. Structural equation model coefficients and associated statistics for Fig. 3.
Funding
Alzheimer’s Association (#NIRG-12-92090 and #NIRP-12-259245; PR-N)
Alzheimer’s Association (#AACSF-20-648075; TAP)
Brain & Behavioral Research Foundation (#29486; DTL)
Brain Canada Foundation (CFI Project 34874; 33397; PR-N)
Canadian Consortium of Neurodegeneration and Aging (#MOP-11-51-31 - team 1; PR-N)
Canadian Institutes of Health Research (#MOP-11-51-31; RFN 152985, 159815, 162303; PR-N)
CNPq (200691/2021-0; JPF-S)
CNPq (166407/2020-8; DTL)
Fonds de Recherche du Québec – Santé (Chercheur Boursier, #2020-VICO-279314; PR-N)
Fonds de Recherche du Québec – Santé (FZL)
National Institutes of Health (#R01AG075336 and #R01AG073267; TAP)
Race Against Dementia Alzheimer’s Research UK (#ARUK-RADF2021A-010; MM)
Weston Brain Institute (PR-N)
Author contributions
Conceptualization: JPF-S, FZL, PR-N, and TAP
Methodology: JPF-S, FZL, and TAP
Formal analysis: JPF-S, FZL, CT, and DLT
Investigation: JPF-S, FZL, DTL, JT, CT, BB, PCLF, MM, Y-TW, GP, ALB, NJA, MC, SS, GB, MSK, JS, NR, VP, NMP, TKK, ERZ, PR-N, TAP
Resources: HZ, KB, PR-N
Writing - Original Draft: JPF-S
Writing - Review & Editing: JPF-S, FZL, DTL, JT, CT, BB, PCLF, MM, GP, ALB, NJA, JTO, JBR, AC, OLL, DLT, TKK, WEK, VLV, J-PS, SG, DOS, HZ, KB, ERZ, PR-N, and TAP
Supervision: TAP
Project administration: JPF-S, PR-N, TAP
Funding acquisition: JPF-S, PR-N, TAP
Competing interests
SG has served as a scientific advisor to Cerveau Therapeutics. HZ has served at scientific advisory boards and/or as a consultant for Abbvie, Alector, Annexon, Apellis, Artery Therapeutics, AZTherapies, CogRx, Denali, Eisai, Nervgen, Novo Nordisk, Pinteon Therapeutics, Red Abbey Labs, reMYND, Passage Bio, Roche, Samumed, Siemens Healthineers, Triplet Therapeutics, and Wave, has given lectures in symposia sponsored by Cellectricon, Fujirebio, Alzecure, Biogen, and Roche, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program. KB has served as a consultant, at advisory boards, or at data monitoring committees for Abcam, Axon, BioArctic, Biogen, JOMDD/Shimadzu. Julius Clinical, Lilly, MagQu, Novartis, Prothena, Roche Diagnostics, and Siemens Healthineers, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program. ERZ serves in the scientific advisory board of Next Innovative Therapeutics. All other authors declare that they have no competing interests.
Data and materials availability
The data used in the present work is not publicly available as the information could compromise the participants’ privacy. Therefore, the data from the TRIAD study will be made available from the senior authors upon reasonable request, and such arrangements are subject to standard data-sharing agreements.
Acknowledgments
We acknowledge all study participants and the staff of the McGill University Research Center for Studies in Aging. We thank Dean Jolly, Alexey Kostikov, Monica Samoila-Lactatus, Karen Ross, Mehdi Boudjemeline, and Sandy Li for assisting in the radiochemistry production, as well as Richard Strauss, Edith Strauss, Guylaine Gagné, Carley Mayhew, Tasha Vinet-Celluci, Karen Wan, Sarah Sbeiti, Meong Jin Joung, Miloudza Olmand, Rim Nazar, Hung-Hsin Hsiao, Reda Bouhachi, and Arturo Aliaga for helping with the acquisition of the data. We also acknowledge Simon Jones and Leonidas Chouliaras at the University of Cambridge for discussing the findings. The views expressed are those of the authors and not necessarily those of the NIHR or the Department of Health and Social Care.
References and Notes




