
Abstract
Epidemiological evidence shows that some diseases tend to co-occur; more exactly, certain groups of patients with a given disease are at a higher risk of developing a specific secondary condition. Despite the considerable interest, only a small number of connections between comorbidities and molecular processes have been identified.
Here we develop a new approach to generate a disease network that uses the accumulating RNA-seq data on human diseases to significantly match a large number of known comorbidities, providing plausible biological models for such co-occurrences. Furthermore, 64% of the known disease pairs can be explained by analysing groups of patients with similar expression profiles, highlighting the importance of patient stratification in the study of comorbidities.
These results solidly support the existence of molecular mechanisms behind many of the known comorbidities. All the information can be explored on a large scale and in detail at http://disease-perception.bsc.es/rgenexcom/.
Introduction
Disease comorbidity is defined as the co-occurrence of two or more conditions in the same patient1. Comorbidity incidence increases with age and has a high impact on life expectancy, as it increases patient mortality and complicates the choice of therapies, posing a major problem for patients and health care systems. Accumulating evidence from epidemiological studies indicates that such co-occurrences do not appear randomly and that specific trends are observed, with some diseases co-occurring more than expected by chance2,3. Systematic studies using electronic health records have been performed to analyze comorbidity patterns in a given population where disease co-occurrences are represented by static networks2,3 or network trajectories if their progression over time is considered4. These studies demonstrated the predictive value of comorbidity patterns to determine disease progression and outcome, including mortality risk.
The observed patterns suggest that comorbid diseases might share underlying molecular mechanisms and risk factors, which can be both genetic and environmental, such as drug exposure and lifestyle. Thus, a better understanding of the molecular mechanisms behind comorbidities is a crucial step towards improved prevention, diagnosis, and treatment of these conditions.
Recent studies on disease comorbidities have included molecular information often analyzing pairs of diseases based on shared disease-related genes5. Similar to functionally related genes, disease-associated genes tend to colocalize in the protein-protein interaction network forming disease modules which can aid the identification of novel candidate genes and inform about disease associations, including phenotypic similarity and comorbidities6. In this context, previous work showed that the overlapping gene expression signatures between several Central Nervous System disorders (CNSd) and cancer types could inform about the molecular mechanisms underlying their direct and inverse comorbidities7,8. More recently, we have observed that disease similarity networks based on gene expression profiles can be used to identify known comorbidity relationships8. Although these efforts were able to capture interesting examples, they were unable to recapitulate what is known at the medical level in a systematic manner. The mentioned approaches based on PPIs and microarrays reproduced a very small percentage of the epidemiology, and other networks based on miRNAs9 and the microbiome10 had no capacity for it. Hence, we address the still largely unknown extent to which molecular information can provide a general explanation to disease comorbidities.
Here, we reformulate the problem and we show, for the first time, that gene expression data is definitively able to reproduce medically known disease co-occurrences. To achieve that, we integrate publicly available RNA-seq data sets, which are currently replacing microarrays due to their improved sensitivity, reproducibility, and detection’s dynamic range11,12. We characterize the gene expression signature of human diseases based on differential expression and functional enrichment analyses. Then, we generate a disease similarity network based on the similarities between diseases’ differential expression profiles. Afterwards, we build a stratified similarity network grouping patients of the same disease with a similar expression profile (here called meta-patients), addressing the fact that patients suffering from a given disease present different risks of developing specific secondary conditions, as evidenced by the Danish medical records4. Both networks are able to significantly recapitulate a large proportion (up to 64%) of the medically known comorbidities2, providing a well-defined set of pathways and molecular functions potentially implicated in disease comorbidities, which can be analyzed at http://disease-perception.bsc.es/rgenexcom/.
Results
Gene expression fingerprint of human diseases
First, we collected published studies analyzing human diseases with RNA-seq data. Uniformly processed gene counts were obtained from the GREIN platform13. After quality filtering (see Methods), 58% of the samples were kept, corresponding to 2.705 samples from 62 studies and comprising 45 diseases (Supplementary Data 1).
We performed differential expression analyses to obtain significantly differentially expressed genes (sDEGs) for each disease (see Methods, Supplementary Table 1). As expected, the number of sDEGs positively correlates with the sample size, whereas it does not correlate with the average library size (average number of sequenced reads) of the diseases (Supplementary Fig. 1).

For each disease, Reactome pathways significantly up-and down-regulated were identified using the GSEA method (FDR <= 0.05). Ward2 algorithm was applied to cluster diseases based on the Euclidean distance of their binarized Normalized Effect Size (1s, and -1s for up-and down-regulated pathways). The heatmap shows the dysregulated Reactome pathways (rows) in the diseases (columns), where up-and down-regulated pathways are blue and red colored respectively. Reactome pathways are sorted and grouped by pathway categories (separated by black horizontal lines). Diseases are colored by ICD9 disease category. Diseases with 0 dysregulated pathways are not shown.
To better understand the transcriptomic alterations associated with the analyzed diseases, we performed functional enrichment analyses14 and, focusing on the significantly enriched Reactome pathways (FDR<=0.05)15, we clustered diseases based on their binarized Normalized Effect Sizes (see Methods, Fig. 1 and Supplementary Fig. 2-4).

Pairwise disease correlations were computed based on the Spearman’s correlation of the union of the sDEGs of each pair of diseases. A disease-disease network was built, containing the significantly positive and negative correlations (FDR <= 0.05), where the edge weights in the network correspond to the Spearman’s correlations (see Methods). The heatmap shows the network’s positive and negative disease interactions, in red and blue respectively. White represents pairs of diseases that are not connected. Diseases are colored by ICD9 disease category.

Precision and recall of the (a) Disease Similarity Network (DSN) and the (b) Stratified Similarity Network (SSN) by disease category pairs. The precision is the percentage of interactions in the molecular networks present in the epidemiological network from Hidalgo et al.2 and the recall is the percentage of epidemiological interactions captured by the molecular networks. For the SSN, interactions between meta-patients and diseases were considered (See Methods). Each point in the symmetric matrix corresponds to the subnetwork that results from selecting the interactions between diseases of the indicated disease categories. The area of the circles represents the recall and the color corresponds to the precision. Green and yellow colors indicate higher and lower precisions than the one of the DSN (43.8%), represented in white. Disease pairs without epidemiological interactions present a single black point. (c) Mean precision of each disease category in the SSN. Disease categories are sorted by their mean precision. Green and yellow colors indicate higher and lower precisions than the one of the DSN, represented in the horizontal grey line. (d) Mean recall of each disease category in the SSN. Disease categories are sorted by their mean recall. Green and yellow colors indicate higher and lower recalls than the one of the DSN (46.2%), represented in the horizontal grey line. Black points indicate the mean (c) precision or (d) recall of the disease categories in the DSN. The differences between the values at the meta-patient and disease level are plotted in the base of the bars.
When considering the pathway enrichment (Fig. 1), two main clusters are defined, one containing less enriched pathways than the other. Most ICD9 disease categories have diseases distributed across the branches, pointing to the involvement of some specific shared biological processes in their pathology. Among others, neuronal system- and extracellular matrix (ECM) organization-related pathways are over and underexpressed respectively in mental and nervous system disorders (bipolar disorder, schizophrenia, Huntington’s disease (HD), Parkinson’s disease and autism), as previously described in the literature16,17. On the other hand, most neoplasms present the expected overexpression of pathways related to the cell cycle, DNA repair, DNA replication, ECM, as well as a decreased hemostasis. Nonetheless, alterations related to developmental biology, immune system and signal transduction are generally observed in specific cancer types18–20.
Moreover, diseases of the digestive system like the Inflammatory Bowel Diseases (IBD): Crohn’s disease and ulcerative colitis, as well as the closely related lymphocytic colitis and ulcer, present an overexpression of a broad set of immune system-related pathways (Supplementary Fig. 2a). Specifically, ulcerative colitis, Crohn’s disease and ulcer form a distinct cluster and share pathways mainly related to cell-cell communication, hemostasis, metabolism, and signal transduction (Supplementary Fig 2b-d and 3a). A common overexpression of multiple specific pathways regarding the ECM organization is observed in IBD (Supplementary Fig. 4c), recently described not only as a consequence of the in situ inflammation but also as an active mediator of it21. Among other pathways, ECM processes are also observed and described to be altered in diseases for which IBD is a risk factor, such as ulcer and colorectal cancer22,23.
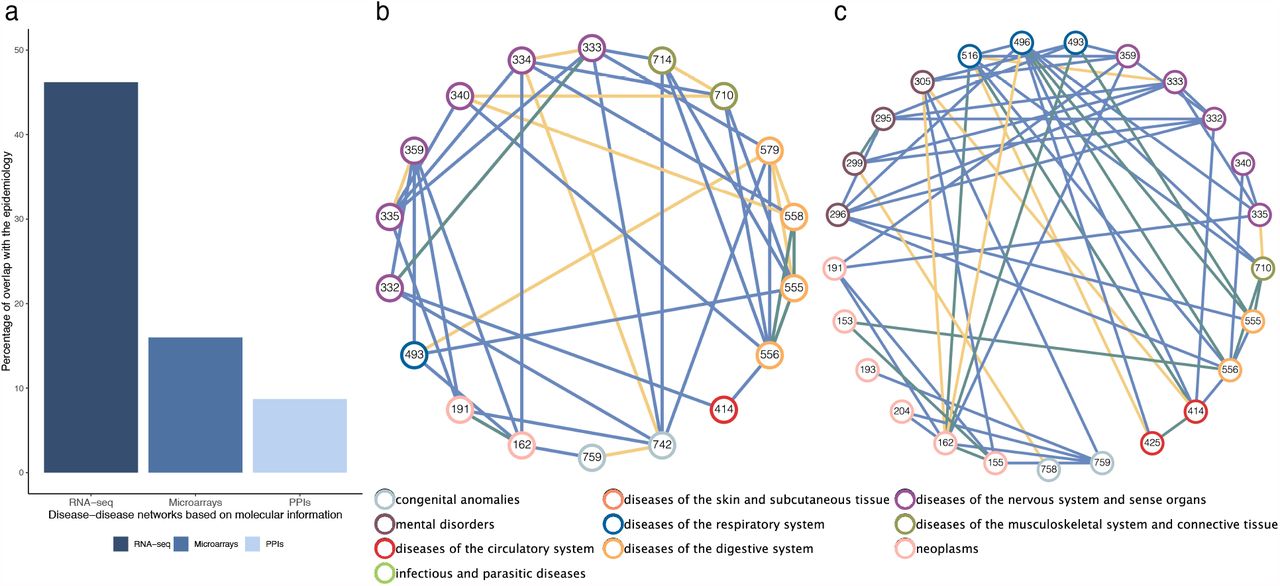
(a) Percentages of interactions in the epidemiological networks, significantly captured by molecular networks. It shows the overlap of our DSN based on RNA-seq and the networks based on microarrays and protein-protein interactions (PPI) (See Methods). (b) Network visualization of the positive disease-disease interactions described in the epidemiological network by Hidalgo et al.2 that captured only by the DSN (blue), only by the network based on PPIs6 (yellow) or by both of them (green). It shows the interactions over the common set of ICD9 codes in both molecular networks. Diseases are colored by their ICD9 code category. (c) Network visualization of the positive disease-disease interactions described in the epidemiological network by Hidalgo et al.2 that captured only by the DSN (blue), only by the DMSN based on microarrays8 (yellow) or by both of them (green).
Disease similarity network
Next, we built a disease similarity network (DSN) connecting diseases based on the Spearman’s correlation (FDR<=0.05) of the genes in the union of their sDEGs (see Methods, Fig. 2). The network is composed of one single connected component -63.37% positive interactions, 36.63% negative interactions -with a mean degree of 29.24 (Supplementary Table 2, Supplementary Fig. 5, see Methods). Nodes’ degree positively correlates with the number of sDEGs and the sample size of the diseases (Supplementary Fig. 6).

Percentage of epidemiological versus non-epidemiological interactions that share (a) overexpressed or (b) underexpressed pathways. Each point represents a Reactome pathway category. The size of the points corresponds to the mean number of shared pathways in the epidemiological interactions.

Percentage of epidemiological (EIs) versus non-epidemiological interactions (NEIs) that share overexpressed pathways. Each point represents a Reactome pathway category. The size of the points corresponds to the mean number of shared overexpressed pathways in the EIs. The color corresponds to the ratio of the mean number of shared pathways in EIs versus NEIs (e.g. red indicates that epidemiological interactions share more pathways than non-epidemiological interactions). The number of EIs and NEIs is indicated between parentheses in the y and x axis labels, respectively. (a) Interactions between congenital anomalies and diseases of the nervous system and sense organs. (b) Interactions within neoplasms.
The DSN captured the previously described IBD comorbidities, as well as intra-disease category comorbidities regarding neoplasms (e.g. lung and liver cancer). We also observed a positive correlation between the differential expression profiles of Kaposi’s sarcoma (KS) and human immunodeficiency virus’ infection (HIV), being this neoplasm one of the most common malignancies in HIV patients as a consequence of their immune deficiency24. We also captured a previously described less frequent link between KS and other diseases characterized by an immune system imbalance, such as IBD25,26.
Many positive interactions entailing diseases of the nervous system and mental disorders are observed, mainly due to shared neurological dysfunction, ECM dysregulation and, in some cases, immune system involvement (Fig. 1 and 2). Among others, schizophrenia was found to be connected to bipolar disorder, autism, and Parkinson’s and Huntington’s diseases, which are known to be comorbid.
Additionally, we observed some negative correlations between the expression profiles of specific central nervous system disorders (CNSd) and cancers27. For instance, HD presents negative correlations with liver, lung, and breast cancer and chronic lymphocytic leukemia, which are known to co-occur less than the expected by chance28. When comparing the altered pathways for both diseases, we find opposite molecular tendencies in multiple of their key pathogenic processes. On one hand, cancer is characterized by an overexpression of cell cycle and gene transcription processes, whereas HD shows increased cell death, apoptosis, mitochondrial dysfunction and a negative regulation of gene expression (Supplementary Fig. 3c-d, 2c and 4a). Kinesins-related pathways, involved in cell division and intracellular transport, are overexpressed in cancer as previously described29, and underexpressed in HD, where its impairment is also characterized30 (Supplementary Fig. 2c). Additionally, immune abnormalities have been extensively described as central to HD and cancerous processes31,32. For instance, we observed an increased interleukin production and signaling regarding Th1-type immune response (e.g. IL-12) in HD, as well as an activation of the complement cascade, being both processes underexpressed in cancer and previously linked to carcinogenesis31–33 (Supplementary Figure 2a).
Subsequently, we evaluated to what extent the DSN is able to capture medically known comorbidities by computing its overlap with the epidemiological network from Hidalgo et al2. We observed that the DSN significantly overlaps 46.2% of the interactions in Hidalgo et al.2 over the common set of diseases (p-value = 0.0018) (Supplementary Table 3, see Methods). We also showed that the overlap of the negative interactions was not significant (p-value = 0.867). The DSN precision and recall varies depending on the disease category pair (Fig. 3a). For instance, diseases of the digestive system present the highest precision (66.4%) with a mean recall of 53.6%. Interactions entailing congenital anomalies are also captured at a high level. On the contrary, highly heterogeneous diseases (e.g. mental disorders) tend to present lower recall values and neoplasms, which often share the dysregulation of multiple pathways without being comorbid, exhibit the lowest precision.
Moreover, we studied the topological properties of the molecular and epidemiological networks (Supplementary Table 4). Both networks contain one single connected component that comprises all the diseases, being the DSN denser. The DSN composed only of positive interactions and the epidemiological network present a transitivity around 0.5, meaning that nodes connected to a third one have 0.5 probability of being connected. Also, both networks present a mean distance below 2, indicating a high connectivity. Then, we compared the topological properties of the positive ICD9-based DSN subnetwork and the epidemiological network, considering only the common set of diseases (Supplementary Table 5). We observe that both subnetworks have a very similar global topology. For instance, they present a significantly equal mean degree and a very similar density and mean distance (slightly higher for the epidemiology) around 0.42 and 1.6 respectively. Although both networks have a similar mean transitivity above 0.5, it is significantly higher for the epidemiological network, possibly due to other forces such as common risk factors or lifestyle. The networks also present a significantly equal mean closeness, betweenness and degeneracy. Finally, we computed the assortativity of the networks labeling the nodes with their ICD9 disease category. We found that both networks present an assortativity around 0; the epidemiological network is slightly depleted in within-category disease links whereas the DSN seems to present a minimal enrichment in those.
Next, we compared our overlap with the ones derived from published disease-disease networks based on other molecular data (see Methods). Both the microbiome10 and the miRNA9 networks yielded non-significant overlaps with the epidemiological network over their respective common diseases and over the common diseases also present in the DSN. The network derived from protein-protein interaction (PPI) networks presented significant yet small overlaps with the epidemiology (8.71% for the entire network and 18.52% over the diseases in the DSN), and the one generated by Sánchez-Valle et al.8 using microarrays presents a significant overlap of 16% with the epidemiological network from Hidalgo et al.2 (Fig. 4a and Supplementary Table 6).
Finally, we compared the networks that present a significant overlap with the epidemiology (PPI and microarray-based networks) with the DSN (Supplementary Table 7-8). The network derived from PPIs and the DSN share 19 ICD9 codes and only 6 out of the 20 interactions present for these diseases in the former are found in the later (there is no significant overlap between them) (Supplementary Table 7-8). Between these common diseases, 29 epidemiologically known comorbidities are connected only in the DSN whereas 10 are unique to the other network (Fig. 4b). The entire DSN provides information for 22 new ICD9 codes and uniquely captures 149 disease links described in the epidemiology.
On the other hand, the microarrays’ network contains 92 ICD9 codes, where 27 of them are analyzed in the DSN. We computed the overlap of both networks over the common set of ICD9 codes, yielding a significant overlap (p-value = 0.027) of 47.02% of the microarrays’ network. Specifically, positive interactions have a significant overlap (p-value = 0.002) of 62.22% whereas the overlap of the negative interactions is not significant (p-value = 0.624) (Supplementary Table 8). Among these common diseases, the DSN yielded 42 new positive interactions that are described in the epidemiological network by Hidalgo et al.2 (e.g. Crohn’s disease and ulcerative colitis) (Fig. 4c). Additionally, the DSN provides information for 14 new ICD9 codes and captures 141 new interactions that match known comorbidities.
Molecular mechanisms behind comorbidities
Once confirmed that the DSN captures a significant percentage of comorbidities, we inspected which are the molecular mechanisms underlying its epidemiological (EIs) and non-epidemiological interactions (NEIs), i.e. interactions for which it was not possible to find a correspondence with the current medical data. We observe that the Reactome pathway categories behind most interactions tend to display a wider range of dysregulation than those affected only in some interactions; i.e. they share a higher number of altered pathways (Fig. 5). Impressively, 95.2% of the EI in the DSN share at least one -and a mean of 21.2-overexpressed immune system pathways, followed by pathways related to the ECM, metabolism of proteins, metabolism and signal transduction, all involved in over 90% of the interactions (means = 10.9, 10.1, 6.3, 16.1). The underexpressed pathways involved in most of the EIs are related to the metabolism of proteins, signal transduction, metabolism, and developmental biology (means = 4.9, 7.2, 13.5, 2.5).
To adequately detect which pathways are specific to EI, we also take into account the ratio between the mean number of shared over or underexpressed pathways by Reactome category in EIs versus NEIs (Fig 5, Supplementary Fig. 7). Overexpressed circadian clock pathways explain more EIs than NEIs, being the mean number of shared pathways 1.7 times higher in the former than in the latter. Digestion and absorption and protein localization are 3.8 and 1.6 times more commonly underexpressed in the EIs.

(a) Heatmap showing breast cancer patients’ similarity and classification through PAM algorithm. The similarity has been defined as 1 – Spearman’s correlation of the gene expression values. Orange represents a positive correlation between the expression profile of the patients whereas blue represents a negative correlation between them. Patients are colored based on their molecular subtype. Patients are divided in 3 subtypes: ER-(Estrogen negative) in green, TNEG (Triple negative patients) in blue and ER+ (Estrogen positive) in red. Patient’s clustering is marked on the left side of the figure. (b) Percentages of interactions in the epidemiological networks, captured by molecular networks in a significant manner. It shows the overlap of the interactions at the meta-patient and disease level based on RNA-seq and the networks based on microarrays and protein-protein interactions (PPI) (See Methods). (c) Interactions between breast cancer disease and its meta-patients with human diseases. Positive interactions are represented in red and negative interactions in blue. Dashed-faded lines represent interactions at the disease level and shared by all the meta-patients whereas solid lines represent meta-patient specific interactions.
The color corresponds to the ratio of the mean number of shared pathways in epidemiological versus non-epidemiological interactions (e.g. red indicates that epidemiological interactions share more altered pathways than non-epidemiological interactions).
Higher resolution results can be obtained by considering interactions between pairs of disease categories. For instance, the shared overexpression of circadian clock pathways seems to be highly specific of EIs entailing CNSd (Fig. 5 and 6a). Actually, the pivotal and putatively causal role of the circadian system in CNSd and their comorbidities has recently been proposed34–36. Although each individual comparison has its particular portrait of the mechanisms underlying disease interactions, some general patterns become apparent. We observe that pathways tend to cluster according to their ability to explain EI versus NEIs. In Fig. 6a we can see a cluster of pathways whose dysregulation is shared by all EIs whereas smaller clusters are mostly present in NEIs. Interestingly, pathway categories involved in more EIs than NEIs also present a higher number of pathways commonly dysregulated in EIs than in NEIs (Fig. 6a, within the upper cluster, redder dots are observed at the left side). In summary, EIs have been found to present more shared altered pathways than NEIs. In fact, if we remove neoplasms, EIs share the alteration of 53.1% and 56.8% more over and underexpressed pathways, respectively. Nonetheless, by inspecting the interactions between neoplasms, we can discern between the mechanisms that are potentially responsible for their common cause from the ones that are more likely due to the convergence of some common functions (i.e. overexpression of pathways related to the immune system or cell-cell communication as well as the underexpression of developmental biology or DNA repair processes) (Fig. 6b and Supplementary Fig. 8). Comorbid neoplasms tend to share a higher number of overexpressed pathways related to organelle biogenesis and maintenance, chromatin organization or cell cycle and an underexpression of pathways such as: metabolism, transport of small molecules or the immune system. Interestingly, around 30% of comorbidities within neoplasms share a highly specific overexpression of protein localization pathways.
Defining disease meta-patients
It has been shown that patients suffering from a given disease often present different risks of developing specific secondary conditions4. We hypothesize that such differential risks might be driven by the existence of disease subtypes. To evaluate it, we studied disease similarities in a stratified manner by applying clustering algorithms to obtain subgroups of cases with a similar expression profile for each disease (see Methods). We consider those subgroups as meta-patients. To evaluate our approach, we selected breast cancer, a disease with known molecular subtypes for which we have two independent studies. The first study contains 20 estrogen positive (ER+) samples and 18 triple negative (TN) and the second study entails 9 estrogen negative (ER-) samples and 9 ER+. Our two independently obtained clusters (using PAM37 and Ward238 clustering algorithms separately) yielded similar results, being PAM the most accurate when grouping cases according to their defined molecular subtypes (Supplementary Fig. 9 and Supplementary Table 9). Breast cancer patient’s clustering and pairwise similarity shows that PAM clustering classifies most of the cases correctly into estrogen negative (ER-), triple negative (TN) and estrogen positive (ER+) (Fig. 7a), even grouping cases with shared molecular subtypes that belong to two independent studies.
Stratified Similarity Network
Since meta-patients are groups of patients from a given disease, they can be treated as any phenotype to which we can apply gene expression analysis methods that assign a significance to their conclusions. Hence, and first of all, we characterized the obtained meta-patients in terms of their differential expression profiles (see Methods). Then, we built a network based on the gene expression similarity between all the meta-patients and analyzed diseases following the methodology described for the generation of the DSN (see Methods). The resulting Stratified Similarity Network (SSN) contains three types of interactions: (1) the previously described disease-disease interactions, (2) interactions connecting different meta-patients and (3) interactions connecting meta-patients to diseases. The SSN can be fully explored in the web application (see Methods) and its topological properties can be found in the Supplementary Table 4. Since the SSN has a considerably higher number of nodes, we observe a large increase in mean degree with respect to the DSN. As captured by the moderate increase in density and mean transitivity, the meta-patients have added some new interactions that were missed by the DSN, being able to uncover new transitive relationships. These properties, along with a concordant decrease in diameter and mean distance, occur across the entire network and subnetworks entailing positive or negative interactions. In fact, by defining meta-patients we significantly increase the number of diseases linked to each disease by 7.64 diseases (p-value=1.596e-10) and 7.29 (p-value = 6.159e-15) for the positive and negative interactions respectively (Supplementary Fig. 10a). We confirmed that this increase in detection power is significant compared to randomly generated meta-patients (p-value = 0 for positive and negative interactions) (see Methods) (Supplementary Fig. 11).
We inspected the links between breast cancer disease and its meta-patients with the rest of diseases (Fig. 7c). We observe that while most of the interactions are shared between breast meta-patients and the disease, several interactions are specific to some of them. For instance, a negative interaction with multiple sclerosis is only found in ER+ and ER-meta-patients while a positive interaction with autism and bipolar disorder is specific to TN and ER-meta-patients.
While breast cancer meta-patients share most of their connections, this is not the case for all the diseases. Actually, the percentage of positive and negative links shared by all the meta-patients from a given disease varies greatly (Supplementary Fig. 10b). For example, CNSd (e.g. schizophrenia, bipolar disorder, MS or autism), known to be highly heterogeneous, show little consistency in their meta-patients’ interactions. On the other hand, neoplasms -that tend to present a consistent and high alteration of multiple biological processes -seem to present a higher correspondence in their meta-patients’ connections.
To evaluate the ability of meta-patients to uncover new comorbidities, we computed the overlap of the interactions between meta-patients and diseases with the epidemiological data (see Methods). Remarkably, meta-patients significantly captured 64.1% (p-value = 0.0187) of the interactions in the epidemiological network from Hidalgo et al.2, which corresponds to an increase in recall of 17.9% with respect to the DSN with a very small decrease in precision of 0.7%. On the contrary, negative interactions do not show a significant overlap (p-value = 0.8035) (Supplementary Table 10). As Fig. 3 shows, most disease categories benefit from the stratification of diseases, since they tend to present a considerable increase of the recall which is usually accompanied by a slight decrease in precision. Aligned with previous results, highly heterogeneous diseases (nervous system and mental disorders) present some of the highest increases in recall (up to 30%); strikingly, also gaining precision. More moderately, this tendency also occurs for circulatory diseases. Respiratory diseases, which often display a wide range of immune system responses, present 10.9% more recall with the same precision than in the DSN.
Finally, we developed a web application (http://disease-perception.bsc.es/rgenexcom/) in which the networks and their underlying molecular mechanisms can be easily inspected (see Methods).
Discussion
So far, many molecular representations of disease interactions have failed to explain a noteworthy number of the medically known comorbidities, being unable to answer the long-standing question of the molecular origin of comorbidities. The generated networks based on RNA-seq profiles provide a convincing and comprehensive answer to this matter, being able to significantly capture and meaningfully explain 64% of the known comorbidities. Hence, they render a qualitative difference over previous studies, providing a solid support to the key role of molecular mechanisms behind comorbidities in a generalized manner.
Actually, the DSN and the epidemiological network are very similar from a topological perspective. They present significantly equal mean degree, closeness, betweenness and degeneracy, as well as very similar density and mean distance; indicating a general resemblance in their overall structure and information flow. They also show close to zero assortativities with respect to disease categories (minimally negative in the epidemiology and positive in the DSN), which implies a slight tendency of diseases from the same category to be linked more than expected in epidemiology. For instance, while the observed molecular similarities connect some neoplasms that are indeed comorbid (e.g. liver cancer with glioblastoma, lung, or colorectal cancer), they also link specific cancers that are not epidemiologically linked. This evidence shows that the presence of shared molecular mechanisms does not always translate into an increased relative risk that is observed in the currently limited medical data. However, we can discern between the mechanisms behind well-established comorbidities from the ones that may be a consequence of an overall molecular similarity, which is especially relevant for neoplasms that share similar dysregulated pathways. Indeed, without considering neoplasms, EIs tend to share over 50% more altered pathways than pairs of diseases without clear evidence of a medical relation (NEIs).
Several ways in which shared molecular mechanisms can underlie direct comorbidities have been proposed. Essentially, molecular mechanisms can be causally or consequentially altered in a given disease; which, in turn, can contribute to the development of a secondary condition. Thus, molecularly based comorbidities can be explained by the following scenarios: (1) both diseases share the same or correlated causal alterations, (2) the molecular mechanisms altered as a consequence of one disease are associated to the second condition or (3) there is a third condition that increases the risk of developing both of them1. These schemes are not mutually exclusive and can be combined in complex manners. In fact, the study of direct comorbidities in longitudinal studies has shown disease trajectories that can be explained by an underlying aggravation and accumulation of specific molecular processes, especially in a chronic manner39. This is the case for the discussed progression of IBD into colorectal cancer40 and for the highly prevalent disease trajectory that has recently been called metabolic syndrome, including obesity, insulin resistance, diabetes, cardiovascular disease or even cancer41. These observations supported the central role of the underlying molecular mechanisms in the study of individual diseases and disease comorbidities, to the point where efforts have already been destined to redefining diseases by incorporating both their clinical features and molecular profiles42.
Comorbidity relationships can be better understood if disease subtypes and patient-specific patterns are taken into account4. Indeed, previous epidemiological studies have identified comorbidities that depend on the disease subtype43–45. In line with this, we introduced the concept of meta-patients and the stratified exploration of their molecular similarities with diseases. The definition of meta-patients unraveled a significant mean of around 14 new subgroup-specific disease connections per disease, increasing the detection power of disease similarities (Supplementary Fig. 10a). This subclassification of diseases based on similarities of the patients’ gene expression profiles can be related to epidemiological observations of comorbidities that depend on patients’ characteristics. In the case of breast cancer, we observed that although the three meta-patients share most of the disease interactions, some specific and interesting ones emerge. For instance, TN and ER-meta-patients are the only ones presenting a positive interaction with autism and bipolar disorder (Fig. 7c). While several studies46,47 have found no significant correlations, recent molecular and epidemiological evidence suggests cancer as a comorbidity of autism48,49. Besides, an enhanced cancer risk has been described for bipolar disorder patients in both genders, being the risk for breast cancer higher but non-significant50. Additionally, we observed a negative interaction between breast cancer, ER+ and ER-meta-patients with multiple sclerosis (Fig. 7c). Again, opposite tendencies are described in the literature for this connection, where the order of appearance of the disease seems to drive the comorbidity pattern. It has been shown that breast cancer patients are 45% less likely to develop multiple sclerosis51. On the other hand, multiple sclerosis patients have been shown to have a significantly increased risk of breast cancer, presumably driven by immunosuppression derived from the associated treatment52. Therefore, our analysis provides new evidence on subgroup-specific comorbidities with a potential molecular explanation. Moreover, we showed that the percentage of recapitulated epidemiological interactions increased from almost half to 64.1% when considering the interactions between diseases and meta-patients, with a slight decrease in precision of 0.7%.
As shown, the generated networks provide the first systematic translation of disease comorbidities into molecular patterns. Previous efforts based on disease-associated genes in a PPI network were limited by the incompleteness of the interactome and the biased knowledge of disease-associated genes for highly studied diseases6. Furthermore, networks based on other molecular sources such as the microbiome10 and miRNAs9 do not overlap epidemiological interactions significantly.
There are at least three factors that allowed us to significantly improve the explainability of known comorbidities captured with gene expression information. First, the better quality and coverage of RNA-seq data, that unbiasedly increases the number and quality of features whose similarity can be compared between diseases. Secondly, an improved methodology based on the existence of a significant correlation between the differential gene expression profiles of each disease pair. As a result, our disease links are robust, stable and independent of the rest of diseases, even if the disease universe changed; contrary to previous attempts based on relative molecular similarities, where disease links depended on the rest of the network8. Finally, the stratification of diseases into subgroups of cases named meta-patients. Opposite to patient-centric approaches, meta-patients can be methodologically treated as phenotypes are handled in gene expression studies; thus, a significance can be associated to their altered genes, pathways and, importantly, disease interactions in the SSN.
Still, there are a number of issues that, if addressed, could improve the quality of the results and the coverage of the comorbidity space. First, the generated networks contain both positive and negative interactions potentially representing direct and inverse comorbidities. While it is possible to validate the positive ones, it is more difficult to validate the less abundant -but equally interesting-negative ones (36.63%) since they are not systematically described in large studies and are only sporadically addressed in the literature53. Nonetheless, we detected known inverse comorbidities such as: HD and specific cancer types or Parkinson’s disease and rheumatoid arthritis54. Therefore, a current limitation in this study is the lack of epidemiological networks entailing inverse comorbidity relationships.
Another limitation is the lack of sample information, such as age, sex, or treatments, which may drive transcriptomic differences between patients. Also, our samples belong to published studies focused on a specific disease in a given tissue (e.g. brain, liver or blood). Since we have cases and controls for each study and disease, we were able to correct for the tissue effect when generating sDEGs at the disease and meta-patient level. However, it would be optimal to have comprehensive data sets of diseases from the same tissue or an array of interesting tissues. Moreover, better defined and annotated disease subgroups as well as their differential comorbidities could help us refine the definition of meta-patients and increase their power to capture their epidemiological associations. We observed that patient stratification is specially important for highly heterogeneous diseases. While some diseases (e.g. breast cancer) showed few links specific to some meta-patients, more heterogeneous diseases (e.g. CNSd like schizophrenia or bipolar disorder and immune system disorders like asthma) present a majority of meta-patient specific links (Supplementary Fig. 10b).
Future perspectives include increasing sample size, so sex-specific disease similarities can be extracted and compared to their epidemiologically described disease interactions55. Furthermore, the molecular coverage of disease comorbidities could be improved by considering other molecular information that may underlie comorbidities within an integrative approach56. For this, large disease cohorts comprising different kinds of omics as well as clinical information would be needed. Furthermore, the obtained molecular similarities could be used to guide drug repurposing and development57.
In summary, we built disease similarity networks based on transcriptomic information that, for the first time, capture and meaningfully explain a sizable percentage of medically known comorbidities in a significant manner. This supports the idea that disease comorbidities have a strong molecular component that is better captured with gene expression profiles than with other molecular sources. Actually, differential gene expression profiles portray the diseases’ altered state in a rich manner since its signal might reflect from genetic alterations to the epigenetic influence on gene expression due to internal or external factors such as treatments, contaminants or lifestyle.
This study shed light into the biological processes underlying known disease comorbidities, leading to a better understanding of the molecular profile and etiology of diseases and their comorbidity relationships. Importantly, although we showed that many of these mechanisms have already been validated experimentally, our efforts propose numerous key genes and pathways that are still to be explored. Thus, we focused our discussion on some examples and provided the molecular characterization of all the diseases and meta-patients at the different levels of granularity (genes and pathways) within a framework that allows for the comparison of the molecular profiles for direct and inverse comorbidities in a detailed and user-friendly manner (http://disease-perception.bsc.es/rgenexcom/).
Finally, our study stresses the need to integrate the study of disease comorbidities and their underlying molecular similarities within a personalized medicine scope, with the aim to capture those disease interactions that might depend on the disease subtype or other patient-specific factors. This would allow us, not only to better understand the putative secondary conditions of specific patients, but to better characterize the underlying molecular processes that might explain those relationships.
Methods
Gene Expression Analysis
Uniformly processed RNA-seq gene counts were downloaded from the GREIN platform13 for 72 human diseases analyzed by 107 studies, including a total number of 4.267 samples.
An RNA-seq pipeline destined to the parallel processing of a collection of RNA-seq studies for a given set of diseases was developed (Supplementary Fig. 12). First, samples with a percentage of aligned reads to the genome lower than 70% were removed, as well as studies with less than 3 cases (from now on patients) and control samples meeting the mentioned requirement. Secondly, and in order to perform the analysis at the disease level, gene counts and metadata for each disease were integrated (only studies with the disease, tissue and disease state information were considered). We performed quality controls using the edgeR pipeline58 and we applied within-sample normalization by considering the logarithm of the counts-per-million (log2CPM). Afterwards, we filtered out lowly expressed genes (those with less than 1 log2CPM in more than 20% of the samples) and we applied between-sample normalization using the trimmed mean of M values (TMM) method59. After performing batch effect identification, we used the limma pipeline60 for differential expression analysis. Specifically, we built a model considering sample type (case vs. control) as our outcome of interest and adjusting for the study effect, as it is the most descriptive independent variable (tissue, platform and others depend on the study of origin). Genes with an FDR <=0.05 were considered significantly differentially expressed genes (sDEGs). Moreover, we used Combat61 and QR Decomposition60 batch effect removal methods to check the clustering of the samples with t-Distributed Stochastic Neighbor embedding (tSNEs)62.
Functional enrichment analysis
In order to better characterize the molecular processes underlying the analyzed diseases, we performed Gene Set Enrichment Analyses (GSEA)14 on the ranked lists of genes based on the differential expression -log Fold Change (logFC)-results using annotations from Reactome15, Kyoto Encyclopedia of Genes and Genomes63 and Gene Ontology64. We considered gene sets and pathways with an FDR <= 0.05 to be significant.
To facilitate the interpretation of the molecular processes altered in diseases and potentially involved in disease comorbidity relationships, we selected the Reactome pathways15 significantly enriched in each disease and applied Ward2 algorithm38 to cluster diseases based on the Euclidean distance of their binarized Normalized Effect Sizes (1s and -1s for up-and down-regulated pathways) (Fig. 1).
Disease similarity network
To define disease-disease similarities we computed, for each disease pair, the Spearman’s correlation between the logFC values of the genes in the union of their sDEGs. We kept the interactions between different diseases that were significant after correcting for multiple testing (FDR <= 0.05). The resulting disease similarity network (DSN) contains positive and negative interactions (significantly positive and negative correlations respectively).
Then, we evaluated the overlap of the obtained positive interactions extracted from diseases’ gene expression similarities with the ones described by Hidalgo et al.2 (based on medical records). To do so, we transformed our disease names into the International Code of Diseases, version 9 (ICD9 codes). Since some diseases share the same three-digits ICD9 code (e.g. muscular dystrophy, myotonic dystrophy and facioscapulohumeral dystrophy share the code 359 -muscular dystrophies and other myopathies), we grouped their samples together and ran the whole analysis (gene expression analysis and disease-disease network building) on them, generating an ICD9 similarity network. Next, we computed the overlap as the percentage of interactions of the epidemiological network -entailing common diseases-captured by the ICD9 DSN’s positive and negative interactions independently. To show the enrichment of our network in epidemiological interactions, we also computed the overlap in the opposite direction. That is, the percentage of interactions in the ICD9 DSN contained in the epidemiological network. We assessed the significance of the overlaps by shuffling the interactions while preserving the degree distribution. We also computed the overlaps directly from the DSN (Supplementary Notes, Supplementary Table 11).
Finally, we compared our overlap with the one obtained with other disease-disease networks based on molecular data. We downloaded networks that link diseases based on the similarities of their microbiome10 and miRNAs9 and we generated a disease-disease network based on protein-protein interactions (PPIs) by selecting the disease pairs that present a significant overlap of their network modules as described by Menche et al.6. Next, we computed their overlap with the epidemiological network over their common set of diseases and over the common set that is contained in our disease set. Finally, we compared the ICD9-level DSN with the networks that significantly overlap the epidemiology (the network based on PPIs and the microarrays’ disease molecular similarity network by Sánchez-Valle et al.8).
Meta-patients generation
We stratified diseases into subgroups of patients with similar expression profiles (meta-patients) by applying clustering algorithms to the normalized and batch effect corrected gene expression matrix. Both PAM (k-medoids)65 and Ward238 algorithms were applied independently (Supplementary Fig. 12).
In the k-medoids approach, we calculated pairwise distances as 1 -the Spearman’s correlation. To obtain the diseases’ meta-patients, first we obtained the optimal number of clusters for each disease by running k-medoids for a cluster number between 2 and 15. After that, the cluster number with the highest average Silhouette value was used to obtain the final meta-patients66.
To evaluate our approach, we selected breast cancer, a disease with known molecular subtypes and for which we have two independent studies. We compared our two independently obtained clusters with the defined disease subtypes (Supplementary Table 9).
Stratified Similarity Network
To analyze the disease subtype-associated comorbidities, we built the Stratified Similarity Network (SSN) connecting meta-patients and diseases based on the pairwise Spearman’s correlation of the union of their sDEGs. First, meta-patient’s gene expression analysis was performed using the same approach described for the diseases, where all the samples corresponding to a given meta-patient were compared with all the controls for the disease. Then, the SSN was built following the same methodology described for the DSN by treating meta-patients and diseases equally as phenotypes. To assess if the meta-patients increase the detection power significantly, we generated 1000 random meta-patients for each disease by shuffling the cases while maintaining the meta-patients’ number and size. Next, we obtained 1000 SSNs and evaluated if the number of positive and negative interactions in the SSN could be observed by chance. To evaluate if meta-patients capture epidemiologically known associations with diseases, we selected the positive interactions between meta-patients and diseases, transformed them into ICD9 codes and computed their overlap with the epidemiological network from Hidalgo et al.2, as described for the DSN. This is comparable to the available epidemiological network, that comprises interactions at the disease level by evaluating if a group of patients from a given disease is at a higher risk of developing a specific secondary condition.
Web application
To facilitate the visualization and exploration of the generated networks, we implemented a web application that displays the DSN and SSN in a dynamic manner67. The user can filter the networks by the type of interactions (positive or negative) and by selecting a minimum and maximum threshold for the edge’s weight. Community detection algorithms (greedy modularity optimization68 or random walks69 can be applied to the filtered network and interactions involving specific nodes can be filtered and highlighted. Furthermore, the molecular mechanisms behind diseases and disease interactions can be easily inspected and compared.
Data Availability
The code of the experiments is available at https://github.com/beatrizurda/Urda-Garcia_et_al_2021 and the code of the web application can be found at https://github.com/bsc-life/rgenexcom. The data is publicly available (Supplementary Data 1); the raw data can be downloaded from GEO (https://www.ncbi.nlm.nih.gov/geo/) and the counts can be downloaded from the GREIN platform (http://www.ilincs.org/apps/grein/).
https://github.com/beatrizurda/Urda-Garcia_et_al_2021
https://github.com/bsc-life/rgenexcom
Data Availability
The code of the experiments is available at https://github.com/beatrizurda/Urda-Garcia_et_al_2021 and the code of the web application can be found at https://github.com/bsc-life/rgenexcom. The data is publicly available (Supplementary Data 1); the raw data can be downloaded from GEO (https://www.ncbi.nlm.nih.gov/geo/) and the counts can be downloaded from the GREIN platform (http://www.ilincs.org/apps/grein/).
Contributions
B.U.-G, J.S.-V., R.L. and A.V. designed all the experiments. B.U.-G. performed all the experiments and developed the web application. All the authors wrote the manuscript and discussed the obtained results.
Conflict of Interest
none declared.
Acknowledgments
This work has been supported by the Ph.D. Fellowship (PRE2019-090454) and funded by the Spanish Ministry of Economics and Competitiveness (RTI2018-096653-B-I00). J.S.-V. was supported by the PhD fellowship BES-2016-077403.
We thank Vera Pancaldi from the Centre de Recherches en Cancérologie de Toulouse, INSERM and Barcelona Supercomputing Center (BSC); Anaïs Baudot from the INSERM, Marseille Medical Genetics and BSC, Jose Luis Portero Navío from Novo Nordisk; Emre Guney from Hospital del Mar Medical Research Institute and Universitat Pompeu Fabra, and Marta Mele from BSC for their helpful critical comments on the work.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}