
ABSTRACT
With the coronavirus disease 19 (COVID-19) pandemic now deep into its second year, widespread testing for the detection of the causative severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is fundamental. The gold standard reverse transcription quantitative PCR (RT-qPCR) cannot keep up with the high demand alone, therefore alternative diagnostic tests are needed. Here we present ADESSO (Accurate Detection of Evolving SARS-CoV-2 through SHERLOCK Optimisation), an optimised version of the CRISPR-based SHERLOCK (Specific High-sensitivity Enzymatic Reporter unLOCKing) assay. After an extensive validation on 983 clinical samples, we demonstrated that ADESSO has a sensitivity of 96% and a specificity of 100% on extracted RNA, comparable to RT-qPCR. Its performance on unextracted samples still allows the detection of the more infectious 75% of the COVID-19 positive population, making it suitable for point-of-care (POC) testing. Interestingly, our in parallel comparison of 390 matching swab and gargle samples showed consistently lower viral loads in gargle specimens. We also validated ADESSO for the detection of the B.1.1.7 variant and demonstrated that ADESSO is adaptable to any variant of concern in less than one week, a critical feature now that worrisome SARS-CoV-2 variants are spreading all around the world.
INTRODUCTION
Since the beginning of the global pandemic of coronavirus disease 2019 (COVID-19), 170 million confirmed cases, including 3.5 million deaths, have been reported1. COVID-19 is a severe respiratory disease caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)2,3,4. The quick diffusion of SARS-CoV-2 is primarily attributed to the relatively long duration of viral shedding by infected individuals, the viral load dynamics and the lengthy incubation period5. Indeed, the incubation period of SARS-CoV-2 is estimated to be 5-6 days6,7 with a high viral load upon the onset of symptoms8–10, suggesting that individuals with COVID-19 begin viral shedding a few days before symptoms appear11. Further, a significant proportion of infected individuals either remain entirely asymptomatic or only manifest mild symptoms12–14. Since these carriers are still able to transmit the virus, case identification and contact tracing protocols alone remain inefficient11,13,15–18, thus facilitating uncontrolled spread of the virus and leading to the current pandemic situation.
The urgent need for a vaccine has accelerated the development of multiple effective vaccines and more than 1.5 billion doses have already been administered1,19–22. However, even in the most optimistic scenario, it will take some time before we will be able to reap the benefit of a global vaccination campaign23,24. Therefore, complementary efforts to limit the spread of the virus are still essential.
To mitigate viral spreading, many countries adopted extreme social distancing measures, including strict lockdowns25. However, the socio-economic costs for such measures are enormous and the consequences will be long lasting26–28. Therefore, the highest priority has to be given to the development of strategies aimed at ensuring long-term safety through containment and isolation of SARS-CoV-2 positive individuals and, at the same time, allowing a safe restart of businesses and social life29. A recent model of viral dynamics suggests that frequent testing for the identification of viral infections and the isolation of carriers is essential30. Notably, the model indicates that effective screening depends mainly on the frequency of testing and speed of reporting, while only to some extent on test sensitivity30.
The worldwide gold standard diagnostic test for SARS-CoV-2 infection is the reverse transcription quantitative polymerase chain reaction (RT-qPCR). Viral RNA is isolated from nasal swabs, throat swabs or saliva, retro-transcribed into cDNA and specific regions of the viral genome are amplified via quantitative PCR. Multiple primer sets are utilised, allowing for the amplification of different targets with a LoD of 103 viral RNA cp/ml31.
An important limitation of RT-qPCR is the requirement for specific equipment, laboratory infrastructures and qualified personnel. Inadequate access to such resources significantly reduces the frequency of testing. Additionally, PCR testing facilities often require days’ worth of time to report the test outcome, resulting in a long sample-to-result turnaround time. To face these challenges, different rapid tests have been implemented, such as rapid PCR and antigen-based tests. However, since rapid PCR tests still require specific equipment and antigen-based tests have lower sensitivity and specificity32, there is a need for an alternative test that is comparable to RT-qPCR in terms of sensitivity and specificity, but faster and independent of complex instruments.
All these requirements are met by the so-called CRISPR diagnostic (CRISPR Dx) technologies, which comprise multiple tools for rapid, economical, sensitive and specific nucleic acid detection33. The CRISPR system is a bacterial machinery able to recognise and cleave foreign genetic material. Among the CRISPR associated (Cas) proteins, Cas13 and Cas12 are able to specifically bind RNA and DNA molecules, respectively, complementary to the target-binding CRISPR RNA (crRNA). Upon target recognition, the Cas proteins cleave a reporter in trans, which can then be detected via different readouts34–36. These readouts are limited by the amount of detectable target material in the sample of interest. To circumvent this limitation, isothermal amplification methods that do not rely on sophisticated equipment, such as loop-mediated isothermal amplification (LAMP)37 or recombinase polymerase amplification (RPA)38 have been combined with Cas-mediated nucleic acid detection35,39,40. CRISPR Dx technologies were quickly adapted to offer point-of-care (POC) diagnostic tests for the detection of SARS-CoV-2. In about an hour, test results can be read on paper-based lateral flow sticks or by fluorescence detection with portable devices41–48. Despite the high potential of CRISPR Dx technologies, only two of them have received emergency use authorisation from the Food and Drug Administration (FDA), with use restricted to the authorised laboratories49,50. The analysis of their performance on clinical samples is still not adequate enough to grant them approval for use in routine diagnostics, therefore a more extensive study in comparison with RT-qPCR is necessary.
Here we have optimised the Cas13a-based detection platform named “SHERLOCK”39,40 and developed ADESSO (Accurate Detection of Evolving SARS-CoV-2 through SHERLOCK Optimisation) for highly sensitive detection of SARS-CoV-2 directly from patient-derived material. The entire protocol takes one hour, does not require RNA extraction or any specific equipment, is able to detect down to 2.5 cp/μl of SARS-CoV-2 synthetic genome and is low-cost (less than 5€ per test). Throughout our work we extensively evaluated the real diagnostic potential of ADESSO in direct comparison to RT-qPCR and with two different sample collection methods (nasopharyngeal swab (NP) vs gargle of saline). Importantly, all the samples were collected from ambulatory patients presenting minimal or mild symptoms or from people identified as contacts of SARS-CoV-2 infected individuals, representing a part of the population that can potentially remain undetected. Our study showed that ADESSO has a sensitivity comparable to RT-qPCR when applied to purified RNA, while it resulted in a lower sensitivity when performed directly on unextracted samples, yet still being more sensitive than rapid antigen tests32. However, considering that the Ct values across our cohort follow a normal distribution, we can fairly estimate that ∼75% of the entire infected population would be successfully detected by ADESSO on unextracted swabs. Importantly, the 25% portion of infected individuals that ADESSO would miss corresponds to high RT-qPCR Ct values, coinciding with low viral titers and minimal infectiousness11,51. Additionally, we also observed a drop in sensitivity when gargling was used as sampling method for both ADESSO and RT-qPCR. Finally, in less than one week we adapted ADESSO to specifically identify SARS-CoV-2 variants in clinical samples by modifying the primers and crRNAs used for amplification and detection. The adapted ADESSO can identify the presence of a variant within one hour of sample submission, thus eliminating the need for sequencing, while RT-qPCR tests would require an additional day or two on top of the already slower turnaround. Considering the risk posed by the spread of the new SARS-CoV-2 variants, this feature is highly relevant in the current phase of the COVID-19 pandemic and in the near future52.
RESULTS
A SHERLOCK-based assay for SARS-CoV-2 detection in clinical samples
The need for a rapid and sensitive COVID-19 POC test has been and will remain a significant factor to contain the spread of SARS-CoV-2. CRISPR Dx technologies represent a viable option for the development of such a test33. For this reason, we first aimed to reproduce and adapt a Cas13-based molecular detection platform called SHERLOCK (Specific High Sensitivity Enzymatic Reporter UnLOCKing)39 for the detection of SARS-CoV-2 in clinical samples. SHERLOCK is based on two main steps: (a) isothermal amplification of viral RNA via RT-RPA and (b) detection of a specific RNA sequence by Cas13 protein followed by trans-cleavage of a labeled reporter, which can be detected via a lateral flow-based visual readout39 (Figure 1A). During the RT-RPA, RNA is retro-transcribed and amplified to dsDNA using specific primers38 and a T7 promoter is added to the amplicon by including its sequence in the forward primer. This feature is necessary for the Cas13 detection step, where a T7 RNA polymerase transcribes the amplified dsDNA back into RNA, which can be specifically recognised by Cas13 protein in complex with a crRNA complementary to the target. The specific binding between the Cas13-crRNA complex and its target RNA activates Cas13 collateral activity, leading to cleavage of an RNA reporter and generation of a detectable signal. Finally, the resulting signal can be read on a lateral flow-based visual readout. For this readout an RNA reporter flanked by biotin and fluorescein (FAM) is used in combination with anti-FAM antibodies labelled gold nanoparticles used to visualise the reporter. In a negative sample, the reporter is intact and is captured by a line of streptavidin resulting in a first band called “control band”. In a positive sample, the reporter is cut, therefore the first half of the reporter containing biotin is captured by streptavidin, while the other half containing FAM is captured by a second line of antibodies resulting in the appearance of the so-called “test band”. The band intensity ratio between the test band and the control band indicates the portion of reporter that has been cut, which reflects the level of Cas13 activation and thus the amount of target RNA that was detected in the sample (Figure 1A).

A. Graphic of SHERLOCK experimental workflow to detect SARS-CoV-2 in RNA extracted from clinical samples with lateral flow readout. B. SHERLOCK sensitivity on serial dilutions of an IVT fragment of SARS-CoV-2 S and Orf1a genes. C. Comparison of SARS-CoV-2 detection on RNA extracted from 30 clinical samples via SHERLOCK and RT-qPCR (Medical University Hospital Mannheim (RT-qPCR hospital) or CDC 2019-nCoV Real-Time RT-PCR Diagnostic Panel). SHERLOCK was performed on SARS-CoV-2 S gene; RT-qPCR at the Medical University Hospital Mannheim was performed on SARS-CoV-2 E and Orf1a genes; CDC RT-qPCR was performed on SARS-CoV-2 N1, N2 and E genes (CDC N1, N2, E) and human RNase P (CDC Rp) as RNA quality control. T = test band; C = control band; nd = not detected; NTC = non template control.
We first generated SARS-CoV-2-specific guide sequences, purified LwaCas13a protein53 (Figure S1A,B) and tested the system’s detection sensitivity in the absence of a pre-amplification step using serial dilutions of an in-vitro-transcribed (IVT) fragment of the SARS-CoV-2 S gene. As previously published, a sensitivity of 108 aM was observed39 (Figure S1C). We then assessed the sensitivity of our test when combining the Cas13 detection with an RT-RPA pre-amplification step on SARS-CoV-2 genes S and Orf1a, as formerly described54. A 2-fold improvement of the previously published sensitivity was obtained54. Indeed, we detected SARS-CoV-2 gene S at a concentration of 10aM (5 cp/μl) and gene Orf1a at a concentration of 100aM (50 cp/μl) (Figure 1B). This improvement is most likely due to the replacement of ProtoScript II Reverse Transcriptase (RT) with M-MuLV RT, which retains a functional RNase H domain that degrades DNA:RNA hybrid intermediates and thereby improves the efficiency of RT55 (Figure S1D). We then used the set of primers and crRNA for S to conduct a blind test on 30 clinical samples. These samples were NP swabs collected in saline (0.9% NaCl) and they were previously tested for SARS-CoV-2 via RT-qPCR (Roche Cobas System) at the Medical University Hospital Mannheim. The specimens were frozen and transported to our laboratory, where we extracted RNA and performed SHERLOCK in duplicates. Additionally, the CDC 2019-nCoV Real-Time RT-PCR Diagnostic Panel56 was also performed as a control. Using SHERLOCK, we were able to identify all 10 positive samples (Figure 1C, Table S1). Notably, we detected sample 28, which had a very low viral titer (corresponding to a high Ct value). These results demonstrate that SHERLOCK can be used as an alternative method to detect SARS-CoV-2 in RNA extracted from clinical samples.
SARS-CoV-2 direct detection from clinical samples
RNA extraction is a time-consuming, labor-intensive and costly step for COVID-19 diagnosis (Figure 1A) and shortage of RNA extraction kits has been a global issue throughout the pandemic57,58. Different studies have already demonstrated that it is possible to omit it41,43,59,60. Therefore, after demonstrating the high potential of SHERLOCK as a diagnostic test for COVID-19, we attempted to improve our protocol in order to avoid the RNA extraction step, thus making the test faster and cheaper (Figure 2A). First, we compared different lysis methods by treating one known positive sample (sample #30 in Figure 1C and Supplementary File 1) with either QuickExtract DNA Extraction solution (Lucigen, #QE09050), Luna Cell Ready Lysis Buffer (NEB, #E3032) or 5% Triton X 100 (Carl Roth, #3051.3) and incubating it for 5 min at 95°C. We performed the experiment in triplicates for each lysis method and we were able to successfully detect SARS-CoV-2 directly after lysing the sample with QuickExtract DNA Extraction solution and Luna Cell Ready Lysis Buffer (Figure 2B). Lysis with 5% Triton X 100 did not allow SARS-CoV-2 detection, although it was successfully performed in another study59. To improve the sensitivity of our test, we first optimised the amount of RT units and sample input in the RT-RPA step using dilutions of synthetic SARS-CoV-2 genome. We observed the best results with 6 U/μl of RT and 2.5 μl of sample input per reaction (Figure S2A). Additionally, we compared our standard set of RPA primers and Cas13 crRNA for S with other sets designed to target different genes of SARS-CoV-2, namely N and Orf1a. First, we assessed their performance on serial dilutions of a positive sample (sample #6 in Figure 1C and Supplementary File 1) (Figure S2B). Then, we compared the sensitivity of the best two candidates, S and Orf1a, on dilutions of synthetic SARS-CoV-2 genome61. The set of RPA primers and Cas13 crRNA for S remained the most sensitive one (Figure S2C). We therefore selected it for all the following experiments. In order to assess the sensitivity of our test on unextracted samples, we spiked in serial dilutions of SARS-CoV-2 synthetic genome in a negative sample lysed with QuickExtract solution and we performed SHERLOCK on the S gene. We were able to consistently detect 10 cp/μl (Figure 2C). We then proceeded with the evaluation of the diagnostic potential of SHERLOCK on unextracted samples (so-called direct SHERLOCK) by performing a second blind test on 160 clinical samples. Positive samples were considered those which resulted in a band intensity ratio (test band/control band) higher than 0.2. This threshold was defined based on the band intensity ratio obtained in all the negative controls used in this study and the negative samples analysed in Figures 2 and 4 (n = 282 + 467; Figure S3A). Direct SHERLOCK was able to identify 73 out of 93 positive samples resulting in a sensitivity of 78% (Figure 2D,E and Table S1). Importantly, despite an apparent LoD equivalent to Ct 27, samples with lower Ct values and high viral loads resulted in a highly variable band intensity ratio with some being very close to the 0.2 threshold (Figure 2D). For this reason, we decided to proceed with a step-by-step optimisation of the direct SHERLOCK protocol.

A. Graphic of SHERLOCK experimental workflow to detect SARS-CoV-2 in unextracted clinical samples with lateral flow readout. B. Comparison of three lysis methods for direct detection of SARS-CoV-2 in a COVID-19 positive clinical sample (sample #30 in Figure 1C) via SHERLOCK with lateral flow readout. Each lysis method was performed in triplicates. C. Determination of SHERLOCK sensitivity with lateral flow readout on serial dilutions of SARS-CoV-2 synthetic genome spiked in a negative sample lysed with QuickExtract DNA Extraction Solution. For B-C band intensity ratios are shown in the bar plots on the right. T = test band; C = control band; NTC = non template control. D. SHERLOCK performance on 160 unextracted clinical samples with lateral flow readout. Only the band intensity ratios of the COVID-19 positive samples (n = 93) are shown in the bar plot. LoD = Limit of Detection. E. Concordance between SHERLOCK (on unextracted samples) and RT-qPCR (on extracted RNA) for 160 clinical samples (93 positive and 67 negative).

A. Measurement of RNase activity in a swab sample lysed at 95°C for 5 minutes with QuickExtract DNA Extraction Solution enriched or not with RNase inhibitor, Murine, at a final concentration of 4 U/μl. B. Comparison of SHERLOCK sensitivity on serial dilutions of SARS-CoV-2 genome with different reverse transcriptases in presence or absence of RNase H. C. Optimisation of SHERLOCK sensitivity with lateral flow readout by increasing the RPA reagents to detect a false negative sample (#L151, Supplementary File 1). A true negative sample (#L126, Supplementary File 1) is used as negative control. The lateral flow strips whose band intensity ratios are plotted here are shown in Figure S4B. 1xRPA corresponds to the standard amount of RPA described in the original SHERLOCK protocol53 and 5xRPA corresponds to the optimal amount recommended by the manufacturer. D. Confirmation of the improved SHERLOCK sensitivity with 2xRPA compared to 1xRPA on clinical samples with Ct values close to our LoD based on Figure 2 (Supplementary File 1). The lateral flow strips whose band intensity ratios are plotted here are shown in Figure S4C. E. Optimisation of the Cas13 reaction kinetics by increasing the amount of Cas13 protein and crRNA in the reaction with fluorescence readout. The reaction kinetics is evaluated by measuring the fluorescence signal at different time-points. The complete 3-hour analysis is shown in Figure S4D. F. Time-point analysis of the optimised Cas13 reaction in half the volume to determine the shortest incubation time required to detect a positive signal with lateral flow readout. The lateral flow strips whose band intensity ratios are plotted here are shown in Figure S4E. G. Sensitivity of the improved SHERLOCK protocol with lateral flow readout on serial dilutions of SARS-CoV-2 synthetic genome upon integration of all the above-described optimisations. Intensity ratios are shown in the bar plot on the right. T = test band; C = control band. H. Graphic of the experimental workflow of ADESSO to detect SARS-CoV-2 in unextracted clinical samples with lateral flow or fluorescence readout.

A. Schematic of the validation study to assess ADESSO performance for SARS-CoV-2 detection in clinical samples in comparison with RT-qPCR (Tib Molbiol). The COVID-19 status of the samples included in the study was initially determined by RT-qPCR (COBAS). ADESSO was performed on both extracted RNA and unextracted samples with lateral flow readout. The results interpretation for ADESSO was performed without knowledge of the outcome of RT-qPCR. B. ADESSO performance on RNA extracted from swab specimens in comparison with COBAS RT-qPCR. Negative samples by Tib Molbiol RT-qPCR are represented in orange. C. ADESSO performance on unextracted swab specimens in comparison with COBAS RT-qPCR (performed on RNA extracted from swabs). D. ADESSO performance on RNA extracted from gargle (G) samples in comparison with COBAS RT-qPCR (performed on RNA extracted from swabs). Negative samples by Tib Molbiol RT-qPCR are represented in pink. E. ADESSO performance on unextracted G samples in comparison with COBAS RT-qPCR (performed on RNA extracted from swabs). Samples missed by ADESSO with low Ct values (<28) according to COBAS RT-qPCR on RNA extracted from swabs but high Ct values according to Tib Molbiol RT-qPCR on RNA extracted from G samples are represented in dark red. F. Correlation analysis of Ct values obtained with Tib Molbiol RT-qPCR (y axis) or COBAS RT-qPCR (x axis) on RNA extracted from swab specimens. Negative samples by Tib Molbiol RT-qPCR are represented in orange and are excluded in the calculation of the correlation (R). G. Correlation analysis of Ct values obtained with Tib Molbiol RT-qPCR (y axis) and COBAS RT-qPCR (x axis) on RNA extracted from G samples. Negative samples by Tib Molbiol RT-qPCR are represented in pink and are excluded in the calculation of the correlation (R). H. Correlation analysis of Ct values obtained after Tib Molbiol RT-qPCR on RNA extracted from G (y axis) and swab (x axis) samples. Negative swab samples are represented in orange; negative G samples are represented in pink; negative samples both as G and swab are represented in red. All the negative samples are excluded in the calculation of the correlation (R). Samples missed by ADESSO with low Ct values (<28) according to COBAS RT-qPCR on RNA extracted from swabs but high Ct values according to Tib Molbiol RT-qPCR on RNA extracted from G samples are represented in dark red. For panels B, C, D, E only the band intensity ratios of the positive samples are shown (n = 95). Values higher than 1 are plotted as equal to 1 for better visualisation. LoD = Limit of detection.
ADESSO: an optimised and highly sensitive SHERLOCK assay
Considering the results of the detection of SARS-CoV-2 directly from clinical samples (Figure 2), we aimed at optimising the SHERLOCK protocol to develop a direct SARS-CoV-2 diagnostic test that is as sensitive as possible. Therefore, we evaluated alternative reagents and different reaction conditions for each one of the three main steps in SHERLOCK, namely, 1) sample lysis, 2) RT-RPA and 3) Cas13 detection (Figure S4A), to increase both sensitivity and speed of the test. At this stage, we assessed Cas13 activation also via a fluorometer to monitor the speed of the reaction in real time. The fluorescence readout is based on the use of an RNA reporter flanked by a fluorophore and a quencher. Upon Cas13-mediated cleavage of the reporter, the fluorophore is cut from the quencher and its fluorescent signal can be detected by a fluorometer (Figure S4A). First, we measured the RNAse activity in a swab sample collected in saline and lysed with the method selected in Figure 2 (QuickExtract DNA Extraction solution and incubation at 95°C for 5 min). To evaluate RNAse activity, RNaseAlert was added to the sample following lysis and fluorescence was measured to evaluate the corresponding nuclease activity. Notably, addition of RNase inhibitors in the lysis buffer prior to heating was sufficient to inhibit RNAse activity almost completely (Figure 3A). Next, we optimised the RT-RPA step by first comparing different RT enzymes in the presence or absence of RNase H. Once again, M-MuLV shows the best sensitivity (5-2.5 cp/μl) in comparison to ProtoScript II or SuperScript III, while the addition of RNase H leads to an improvement for SuperScript III only (Figure 3B). Secondly, we used different final concentrations of RPA, where 1xRPA corresponds to the standard amount of RPA described in the original SHERLOCK protocol39,53,54 and 5xRPA corresponds to the optimal amount according to the manufacturer’s instructions. To test this, we selected one false negative sample from our previous blind test on unextracted samples (sample #L151, Supplementary File 1) and a true negative sample as negative control (sample #L126, Supplementary File 1) and we repeated our assay with different concentrations of RPA. Remarkably, while the false negative sample is still negative with 1xRPA, it results positive for final concentrations of RPA from 2x to 5x, with a decrease in band intensity ratio when using the 4xRPA and 5xRPA concentrations (Figure 3C, S4B). Considering this and bearing in mind the cost per single test, we decided to proceed using a 2xRPA concentration. To further confirm this improvement, we compared 1xRPA and 2xRPA on 5 samples with Ct values close to the LoD determined in Figure 2 (samples #L95, L96, L111, L122 and L123, Supplementary File 1). We observed an improvement in the 2xRPA reactions with these samples as well (Figure 3D, S4C). Furthermore, in order to optimise the Cas13 detection step we made a ten-fold dilution of a positive RT-RPA reaction (50 cp/μl) and we performed Cas13 detection using the original concentration of Cas13/crRNA (45/22.5 nM)39,53,54, in comparison to higher amounts (Figure S4D, upper panel). A concentration of Cas13/crRNA of 90 nM each leads to an improved reaction, reaching the plateau after 15 min only, compared to 30 min for the other two concentrations (Figure 3E, S4D, lower panel). We also confirmed that a 10-min incubation for Cas13 detection performed in half the volume is sufficient to yield a clearly positive outcome in the lateral flow detection assay (Figure 3F, S4E), which is an essential aspect for a POC test. Moreover, a shorter Cas13 reaction allows us to extend the incubation time of the RT-RPA step for highly sensitive reactions53 without affecting the total time of the assay. Finally, we assessed the sensitivity of this optimised protocol on serial dilutions of SARS-CoV-2 synthetic genome and we observed a robustly reproducible sensitivity of 2.5 cp/μl (Figure 3G). We named this new optimised diagnostic assay ADESSO (Accurate Detection of Evolving SARS-CoV-2 through SHERLOCK Optimisation) (Figure 3H).
Evaluation of ADESSO performance on clinical samples in direct comparison to RT-qPCR
We used ADESSO to test a total of 195 clinical samples in direct comparison to the RT-qPCR protocol routinely used in the clinics. To allow a fair comparison between the methods, we first selected 95 positive and 100 negative individuals (via COBAS RT-qPCR on NP swab). For each of these specimens, RNA was re-extracted and analysed by RT-qPCR (Tib Molbiol) and ADESSO. Additionally, ADESSO was also performed directly on unextracted samples. Finally, we also obtained gargled saline from the same individuals as an alternative sampling method, which would be ideal for POC testing. Those samples were treated exactly as the NP swabs (Figure 4A). Importantly, the ADESSO results were analysed without knowing the outcome of the COBAS RT-qPCR used as the reference.
The results of this experiment are summarised in Table 1. ADESSO on RNA extracted from swabs was able to correctly identify most positive samples (91 out of 95), resulting in a sensitivity of 96% (Figure 4B). Interestingly, all the false negative samples have Ct values higher than 31, corresponding to lower viral loads (<100cp/μl) and therefore a lower probability of spreading the virus11,51. RT-qPCR (Tib Molbiol) performed on the same samples was largely in agreement with the COBAS RT-qPCR, with highly correlated Ct values (Figure 4F). However, using this method we were able to identify 89 out of 95 positive samples resulting in a sensitivity of 94% (Table 1 and Figure 4F). As expected, ADESSO on unextracted samples resulted in a lower sensitivity (77%), with all false negative samples having Ct values higher than 29 (∼100cp/μl) (Table 1 and Figure 4C). The same analysis was performed on gargle saline samples. In this case, ADESSO on extracted RNA correctly identified 74 out of 95 positive samples resulting in a sensitivity of 78%, with most false negative samples having Ct values higher than 30 and few with Ct values between 28 and 29 (Figure 4D). Interestingly, this drop in sensitivity does not seem to be related to the detection method but rather to the sampling method. Indeed, the same decrease in sensitivity (to 79%) was observed also for RT-qPCR (TibMolBio), with true positive samples resulting in higher Ct values (Table 1 and Figure 4G). Finally, as observed for swabs, ADESSO on unextracted gargle saline samples resulted in a lower sensitivity (65%) (Figure 4E and Table 1). In this latter case, false negative samples have different Ct values, with some corresponding to high viral loads in swabs (analysed by COBAS RT-qPCR) (dark red dots in Figure 4E and 4H). The overall decrease in sensitivity is in agreement with the consistently higher Ct values observed in gargle specimens compared to their matched swab samples analysed via Tib Molbiol RT-qPCR (Figure 4H).
Positive and negative predictive values, sensitivity and specificity of ADESSO on swab and gargle samples with and without RNA extraction.
Altogether, these results validate the high potential of ADESSO as a POC test for the detection of SARS-CoV-2 infected individuals. Notably, ADESSO on extracted RNA, either from swab or gargle samples, performed as well as RT-qPCR (Tib Molbiol) in terms of sensitivity and specificity. Additionally, our data also shows an important difference in the detection of SARS-CoV-2 when gargling with saline was used as a sampling method. Even though this approach would be better suited to a POC test, we observed a clear reduction in sensitivity, independently of the detection method used.
Adaptation of ADESSO for detection of SARS-CoV-2 variants: a flexible and powerful assay to rapidly identify specific variants or mutations
Since the beginning of the pandemic, SARS-CoV-2 has evolved considerably. The first variants to appear carried a D614G mutation in the spike protein62, which is now dominant and shared between all the existing variants. While several variants exist, here we focus our attention on two variants of concern: SARS-CoV-2 B.1.1.7 (UK variant) and SARS-CoV-2 B.1.351 (South Africa (SA) variant). SARS-CoV-2 B.1.1.7, also known as 501Y.V1, seems to have an enhanced transmissibility63 and might be more virulent64. It was first detected in England in late 2020 and, after becoming the dominant variant in the UK, it has spread quickly all over Europe and worldwide. B.1.1.7 contains eight mutations in the spike gene in addition to the mutation causing the D614G substitution, including deletions (e.g., ΔHV69-70) (Figure 5A). SARS-CoV-2 B.1.351, also known as 501Y.V2, was first detected in late 2020 in Eastern Cape, South Africa65. This variant quickly became dominant locally and displaced other viral lineages in several regions, possibly as a result of increased transmissibility or immune escape65,66. B.1.351 contains nine mutations in the spike gene in addition to the mutation causing the D614G substitution, including clusters of mutations (e.g., mutations leading to Δ242-244 and R246I) (Figure 5A). Finally, there is growing evidence that these new variants could impair the efficacy of current monoclonal antibody therapies and vaccines because of the several mutations located in the spike gene67–69. For this reason, it is now essential to quickly identify individuals infected by SARS-CoV-2 variants. The UK variant is the major concern in Europe and Germany, therefore we adapted our test to detect the deletion (ΔHV69-70) specific to this strain. We called this adapted test ADESSO-UK (Figure 5A,B). First, we designed two different crRNAs able to recognise either the original Wuhan strain or the UK variant, called respectively crRNA HV69-70 and crRNA ΔHV69-70 (Figure 5B). Then we optimised RT-RPA primers to amplify the region of SARS-CoV-2 genome containing HV69-70 and we selected the more sensitive set 1 for further analysis (Figure S5A). Finally, we performed a blind test on positive clinical samples carrying either UK or SA variants. We first applied ADESSO for the detection of SARS-CoV-2 and we were able to detect all positive samples but one (sample #11, Figure 5C). Then, using ADESSO-UK (crRNA ΔHV69-70 or HV69-70) we were able to correctly identify all the samples carrying the UK variant and we could distinguish the ones bearing the SA strain (samples #1-13, Figure 5D,E and S5B). Interestingly, among the three samples carrying the SA variant, only sample #11 was not detected via ADESSO. Sequencing analysis of the viral genome in these three samples showed that they all shared the deletion Δ242-244, but only sample #11 carried the R246I mutation (Figure S5C). This mutation falls exactly within the bases recognised by the 3’ end of the forward primer used in the RT-RPA step of ADESSO, thus disrupting its function (Figure 5F). Notably, the assay seems to be resistant to deletions of several nucleotides occurring in sequences that are complementary to the central region of the primer (Figure 5F). Altogether, these results show how ADESSO can be readily adapted for the detection of SARS-CoV-2 variants of concern and even specific mutations. The entire adaptation of the test took less than one week, from the selection of a unique mutation for the UK variant to the validation of the adapted protocol, including designing and production of the specific reagents. This feature of our assay is a crucial aspect in the current phase of the COVID-19 pandemic, where quick and sequencing-independent detection of variants is essential to contain their spread52.

A. Schematic of SARS-CoV-2 S gene with annotation of the reported mutations for SARS-CoV-2 B.1.1.7 (top) and B.1.351 (bottom) lineages. The regions of the S gene targeted by ADESSO and ADESSO-UK are indicated in purple and orange, respectively. B. Schematic of the S gene region containing the ΔHV69-70 deletion (highlighted in pink) specific for the B.1.1.7 variant in comparison with the original SARS-CoV-2 sequence from Wuhan and illustration of the binding of the specific crRNAs targeting the mutated (crRNA ΔHV69-70) or Wuhan (crRNA HV69-70) sequence. The grey sequence in the crRNAs is called direct repeat (DR) and its stem-loop structure is needed for the recruitment of Cas13. C. SARS-CoV-2 detection by ADESSO in 13 clinical samples carrying either the UK (B.1.1.7) or SA (B.1.351) SARS-CoV-2 variant. The band intensity ratios are shown in the bar plot on the right. D. SARS-CoV-2 B.1.1.7 variant detection by ADESSO-UK with ΔHV69-70 crRNA and (E) confirmation of the presence of SARS-CoV-2 B.1.351 variant by ADESSO-UK with HV69-70 crRNA in the same samples. For C, D, E, T = test band; C = control band; NTC = non template control. F. Schematic of the binding of the forward RPA primer used in ADESSO to the complementary region in the original SARS-CoV-2 sequence from Wuhan (top), in clinical samples #12 and #13 carrying the SA variant with the deletion Δ242-244 (middle) and in sample #11 carrying the SA variant with an additional mutation (R246I) that disrupts the primer binding (bottom). The positions of the Δ242-244 deletion and the R246I mutation are highlighted in grey. The point mutation causing the R246I substitution is marked in red.
DISCUSSION
The COVID-19 pandemic has been afflicting the world for more than a year now and the number of new weekly global cases is still hitting its highest levels, despite multiple effective vaccines being distributed1. Therefore, promptly tracking infected individuals to isolate them and prevent further spread of the virus is fundamental. The gold-standard RT-qPCR-based COVID-19 diagnostic test alone cannot keep up with the high demand for testing and the long turnaround time is an issue when a fast response is essential. Rapid PCR and antigen-based tests are also available, but there are some limitations for their widespread use, such as the requirement for sophisticated PCR equipment and the standard practice of confirming positive antigen-based test results by RT-qPCR. Therefore, an alternative test that overcomes these limitations is still needed. Here, we have optimised the Cas13a-based diagnostic platform called SHERLOCK39 and developed the improved protocol ADESSO for highly sensitive COVID-19 testing. Overall, we tested 983 samples (496 positive and 487 negative, Supplementary File 1), in parallel comparison with RT-qPCR. To our knowledge, it is the first time that such an extensive study on clinical samples has been reported for CRISPR Dx technologies. ADESSO has a sensitivity of 96% on RNA extracted from swabs and a sensitivity of 77% when performed directly on unextracted swab samples. This drop in sensitivity is due to a decreased LoD at Ct 29, corresponding to low viral titers and minimal infectiousness11,51. However, skipping the RNA extraction step considerably reduces the sample-to-result turnaround time and allows more frequent testing, which is suggested to be essential for efficient identification of viral infections and isolation of carriers to contain the pandemic30. Other advantages are the lower need for RNA extraction kits, whose shortage has been a global issue throughout the pandemic57,58, and the higher test portability, which makes ADESSO appropriate for POC testing. To further increase the POC suitability of ADESSO, we assessed its performance in comparison with RT-qPCR on gargle samples obtained from the same individuals from whom swabs were collected. Interestingly, we observed a loss in sensitivity independently of the diagnostic test. On RNA extracted from gargle samples, ADESSO showed a sensitivity of 78%, comparable with a 79% sensitivity of the RT-qPCR. In line with what we observed for swab samples, the sensitivity of ADESSO decreased to 65% when performed directly on unextracted gargle samples (Table 1). This overall loss of sensitivity can be attributed to the sampling method, which leads to a general increase in Ct values in gargle samples compared to swabs collected from the same individuals at the same time (Figure 4H). To our knowledge, this is the first study that compares two different sampling methods in parallel on such a big cohort of patients (n = 195 swabs + 195 gargle samples), thus highlighting a consistent difference in detected viral titers depending on the sampling method used. Despite the fact that multiple studies have shown that high SARS-CoV-2 titers can be detected in saliva70,71, our results show that using gargle samples instead of NP swabs, even if more suitable for POC testing, leads to higher rates of false negative samples, independently of the sensitivity of the downstream diagnostic test. Therefore, alternative methods should be considered and evaluated in comparison to gargling and NP swabbing, for example self-collection of nasal swabs, whose feasibility and reliability are already being investigated72.
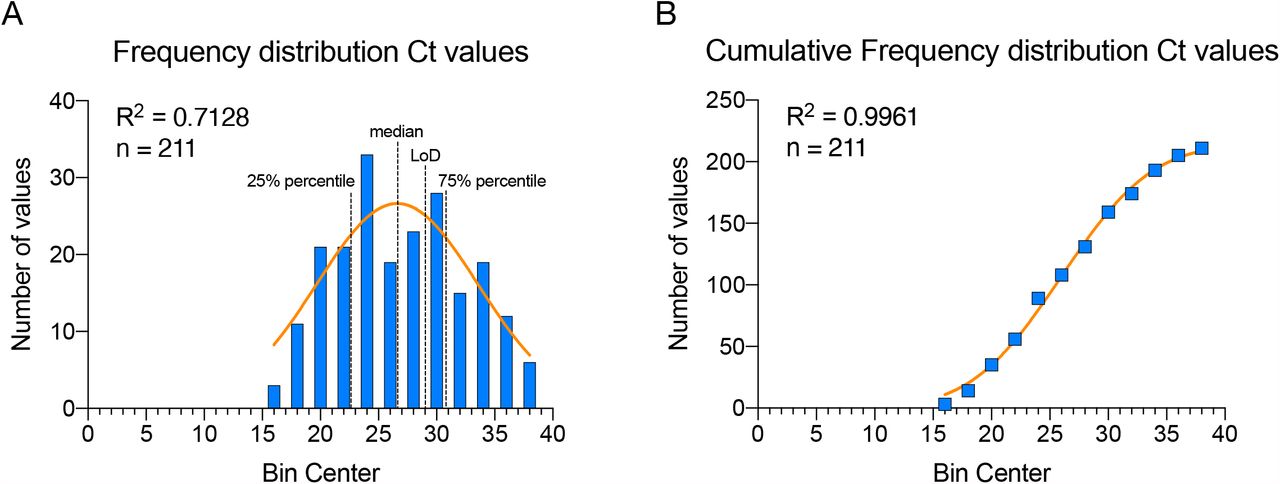
Importantly, the cohort of positive individuals analysed in our study was selected randomly and displays a normal distribution of Ct values, covering the full range of viral titers between Ct 17 and Ct 38 (Figure S6A,B). This aspect is fundamental for two reasons: first, since the samples analysed in this study were collected from ambulatory patients presenting minimal or mild symptoms or from people identified as contacts of SARS-CoV-2 infected individuals, it highlights the fact that these individuals can manifest high viral loads and therefore be infectious; moreover, it allows the inference of the test performance from the experimental cohort to the entire SARS-CoV-2 infected population. In this way, we could confidently estimate what portion of the population our test would detect. Mathematical models show that successful identification and isolation of 50% of infected individuals (and tracing of their contacts) is already sufficient to flatten the infection curve73. Our test exceeds this fraction in all conditions (Table 1), strongly suggesting that immediate and widespread application of ADESSO would be of great help to contain the pandemic. In particular, by applying ADESSO on swab samples without RNA extraction, an estimated ∼75% of the infected population would be successfully detected, while ∼25% of the infected individuals could be missed (Figure 4 and S6A). Importantly, this 25% portion corresponds to individuals with Ct values higher than 29, associated with low viral titers and minimum infectiousness11,51. Finally, our results show a disagreement between LoD on serial dilutions of synthetic viral genome and LoD in clinical samples. Despite the “synthetic” LoD of 2.5 cp/μl (∼Ct 35; Figure 3), the real clinical sensitivity of ADESSO corresponds to Ct 29-31 (Figure 4) and the same is true for other studies although it has never been explicated43,46. This aspect highlights that an extensive validation on real clinical samples covering the full range of viral titers and following a normal distribution, as the one presented here, is necessary to determine the real LoD of a diagnostic test. This is crucial to allow a fair comparison between sensitivities resulting from independent studies, which can be greatly influenced by the choice of the tested population.
The importance of testing is further highlighted by the recent emergence of SARS-CoV-2 variants, which poses a new threat for humanity, as India’s recent tragic crisis has shown52,74. This is of utmost criticality because mutations in the viral genome might impair both molecular and antigen-based tests, thus leading to false negative results. In these situations, being able to promptly adapt a test is fundamental and ADESSO offers such an advantage. Here, in less than a week we adapted the test for the detection of the B.1.1.7 variant. Based on the publicly available SARS-CoV-2 sequences (https://www.gisaid.org), ADESSO can be adapted to any variant of concern, thus providing an all-in-one SARS-CoV-2 detection and variant identification tool without need for sequencing.
Finally, we calculated a cost per reaction of 2.64€ and 4.82€ for fluorometric and lateral-flow detection, respectively (Supplementary File 2), which would be even lower at a production scale. Considering the cost range for a single RT-qPCR reaction75, three to eight tests could be performed with ADESSO for the same price. Moreover, the cost of a thermal cycler needed to perform RT-qPCR would also be eliminated. Lastly, the use of ADESSO for the detection of variants would even cut the cost of sequencing. Altogether, ADESSO is cheaper than any RT-qPCR-based COVID-19 diagnostic test and offers a more accessible option for widespread and more frequent testing.
With the COVID-19 pandemic now deep into its second year, it has become clear that time plays a critical role in the management of such an emergency. In order to control it, rapid detection of new infections, quick tracing of contacts, fast vaccine distribution and prompt reaction to emerging new variants are key-factors. Slowly but undeniably, the race against SARS-CoV-2 has turned from a sprint into a marathon. If we want to keep up, we need to take action faster than the virus evolves. The time is now for ADESSO to join the race.
METHODS
Protocols
The RT-RPA and Cas13 reaction protocols used for each experiment are provided in Supplementary File 4 with reference to the corresponding figures. The exact volumes are given for one single reaction.
Reagents and materials
Detailed information about reagents and material used in this study is provided in Supplementary File 3.
Cas13 purification
Plasmid encoding LwaCas13 (pC013 - Twinstrep-SUMO-huLwCas13a was a gift from Feng Zhang (Addgene plasmid # 90097; http://n2t.net/addgene:90097; RRID:Addgene_90097)39 was transformed into Rosetta cells and purified according to established protocols with substantial modification. Single colonies were inoculated into 25 ml Terrific Broth (TB) (100 μg/ml AMP) and grown to an OD of 0.6 at 37°C degrees while shaking at 150 rpm. The suspension was chilled for 30 min at 4°C and subsequently induced with 0.5 mM IPTG and left shaking for an additional 16h at 21°C. Cells were harvested by centrifugation at 5 k rpm for 15 min at 4°C. The pellet was resuspended in 4x (wt/vol) supplemented lysis buffer (12 cOmplete Ultra EDTA-free tablets, 600 mg of lysozyme and 6 μl of benzoase to lysis buffer (20 mM Tris pH 8.0, 500 mM NaCl, 1 mM DTT)) and lysed by sonication. Lysate was cleared by centrifugation at 10 k rpm for 1h at 4°C. Supernatant was purified using a 1 ml HIS-Trap column (Cytiva) slurry and affinity chromatography was performed using the ÄKTA pure system with lysis buffer for washing steps and an imidazole gradient for elution. After initial purification, the protein sample was incubated with SUMO protease (ThermoScientific) as per the manufacturer’s instructions at 4°C overnight to remove the affinity tags. The sample was then re-applied to a 1 ml HIS-Trap column. Both the SUMO protease (which itself has a 6xHIS tag) and the cleaved affinity tag bind to the resin, while pure Cas13 eluted in the wash step. A final size-exclusion chromatography step was performed using the ÄKTA pure system using 10 mM HEPES pH 7.0, 5 mM MgCl2, 1 M NaCl and 2 mM DTT as gel filtration buffer on a Superdex 16/600 column.
Synthetic SARS-CoV-2 RNA
Fully synthetic SARS-CoV-2 RNA was purchased from Twist Biosciences (MT007544.1 or MN908947.3). In order to test SHERLOCK sensitivity, serial dilutions were prepared in water or in saline, from the initial concentration of 106 cp/μl to 0.01 cp/μl.
Synthetic SARS-CoV-2 S gene and Orf1ab gene RNA fragments
SARS-CoV-2 RNA, a kind gift of Prof. Bartenschlager (DKFZ, Heidelberg), was used for OneStep RT-PCR (Qiagen, #210212) as follows: 11 μl of nuclease-free water, 5 μl of 5x OneStep RT-PCR buffer, 1 μl of dNTP mix (10mM each), 1.5 μl of each primer (forward and reverse, both 10μM) and 1 μl of OneStep RT-PCR Enzyme Mix were added to 4 μl of denatured RNA. The primers used for the amplification of SARS-CoV-2 S gene and Orf1a gene are listed in Supplementary File 3. The RT-PCR protocol was run as follows: retrotranscription at 50°C for 30 min, denaturation at 95°C for 15 min, followed by 40 cycles of denaturation at 94°C for 30 sec, annealing at 61°C (Orf1a gene) or 62°C (S gene) for 30 sec and elongation at 72°C for 5 sec. In the end a final elongation step at 72°C was run for 10 min. PCR clean-up was performed on the RT-PCR products according to the manufacturer’s instructions (Macherey-Nagel, #740609.250). The purified DNA was in-vitro-transcribed into RNA with the HiScribe T7 Quick High Yield RNA Synthesis Kit (NEB, #E2050S) following the suggested protocol for short transcripts. The IVT products were then treated with DNase I (HiScribe T7 Quick High Yield RNA Synthesis Kit, NEB, #E2050S) and purified with Monarch RNA Cleanup Kit (NEB, #T2050). The concentration of the purified products was determined by Nanodrop and Qubit. In order to test SHERLOCK sensitivity, serial dilutions were made in water from a concentration of 1μM to 1aM.
Human clinical specimen collection
Clinical specimens were collected at the Medical University Mannheim, Germany. NP swabs and gargle samples were collected from ambulatory patients presenting minimal to mild symptoms or sent by the German Health Department after having contact with a SARS-CoV-2 positive person. After verbal and visual instruction gargling was performed with 8 ml of sterile 0,9% saline (Fa. Fresenius Kabi, Bad Homburg, Germany). Samples were collected in sterile containers without additives and stored at 4°C until testing with PCR within 36 h. NP specimens were collected with flocked swabs (Improswab, Fa. Improve Medical Instruments, Guanzhou/China) and washed out with 2 ml 0,9% saline within 12 h of collection. For sample inclusion in the validation study and side-by-side comparison of ADESSO and RT-qPCR, initial PCR was performed on NP swab samples as part of routine clinical care using the cobas 6800 system (Roche, Penzberg, Germany) according to the manufacturer’s instructions. Based on the results of the initial PCR, 95 positive and 100 negative samples were selected.
RNA extraction
For the first blind test (Figure 1), RNA was extracted from the clinical samples with the QIAamp® Viral RNA Mini kit (Qiagen, #52904) following the manufacturer’s instructions (140μl of swab were extracted and eluted in 60μl). For the validation study (Figure 4), RNA was extracted from 200 µl of the selected gargle and NP specimens with the MagnaPure Compact System (Roche, Penzberg, Germany) using the Nucleic Acid isolation Kit I (Roche) resulting in 100 µl of eluate. Residual volume of gargle and NP specimens was stored at 4°C and sent to the DKFZ for further analysis.
RT-qPCR
CDC taqman RT-qPCR initially (Figure 1) was performed in technical triplicates according to published protocols76, which we adapted to a 384-well plate format and a reduced reaction volume of 12.5 µl. The reaction was performed using the Superscript III One-Step RT-PCR kit with Platinum Taq Polymerase. Magnesium sulphate and BSA were added to the reaction to a final concentration of 0.8 mM and 0.04 μg/μl, respectively. Primers and probes for the viral N1 and N2 and the human RNase P genes were added as ready-made mix (1 µl; Integrated DNA Technologies Belgium; CatNo. 10006713). The E-gene probes and primers (GATC, Germany) were used at final concentrations of 500 nM for each primer and 125 nM for the probe. ROX was added to a final concentration of 50 nM. PCR was performed in a QuantStudito 5 thermocycler, with cycling conditions 55°C for 10 min, 95°C for 3 min, followed by 45 cycles of 95°C for 15 s and 58°C for 30 s.
For the validation study (Figure 4), real-time PCR of 10 µl RNA-eluate was performed on a BioRad CX96 cycler using the Sarbeco E-Gen-Kit (Fa. Tib Molbiol, Berlin, Germany) following the manufacturer’s instructions. Residual volume of extracted RNA from gargle and NP specimens was stored at −20°C and sent to the DKFZ for further analysis.
Lysis of clinical samples for direct SARS-CoV-2 detection
Clinical samples were lysed for direct SHERLOCK or ADESSO assay (Figures 2) as follows: after vortexing, 10μl of sample were mixed with 10μl of QuickExtract DNA Extraction solution (Lucigen, #QE09050). In the optimised protocol (Figure 4), QuickExtract DNA Extraction solution is enriched with Rnase Inhibitor, Murine (NEB, #M0314) at a final concentration of 4U/μl. Samples were then incubated at 95°C for 5 min. After incubation, samples were mixed by vortexing and spun down for 15 seconds at 10.000g. Finally, 5.6 μl of sample (for RT-RPA 2X) were collected from the upper liquid phase, carefully avoiding to aspirate any precipitate, and used in the RT-RPA step.
crRNA synthesis and purification
CRISPR-RNAs (crRNAs) were either designed in our lab or synthesised by Integrated DNA Technologies (IDT). All crRNAs used in this study are listed in Supplementary File 3. To produce the crRNAs in our lab we followed a previously published protocol53. In short, the templates for the crRNAs were ordered as DNA oligonucleotides from Sigma-Aldrich with an appended T7 promoter sequence. These oligos were annealed with a T7-3G oligonucleotide, and used in an in vitro transcription (IVT) reaction (HiScribe T7 Quick High Yield RNA Synthesis Kit, NEB, #E2050S). The crRNAs were then purified using Agencourt RNAClean XP Kit (Beckman Coulter, #A63987). The correct size of the crRNAs was confirmed on a UREA gel and the concentration evaluated by nanodrop. Aliquots of 10ng/μl of each crRNA were produced to avoid repeated freeze and thaw cycles and stored at −80°C.
Reverse Transcriptase Recombinase polymerase amplification (RT-RPA)
RT-RPA reactions were carried out with TwistAmp Basic (TwistDx, #TABAS03KIT) with the addition of M-MuLV Reverse Transcriptase (NEB, #M0253) and RNase Inhibitor, Murine (NEB, #M0314). Reactions were run at 42°C for 45 minutes in a heat block. Here are the details for the optimised reaction (so called RT-RPA 2X): two lyophilized pellets TwistAmp Basic are used to prepare the following master mix for 5 reactions: 59 μl of Rehydration Buffer (RB) are mixed with 2,5 μl of each primer (forward and reverse) at a concentration of 20μM, 1.5 μl of M-MuLV RetroTranscriptase (200U/μl - NEB, #M0253) and 1,5 μl of Rnase Inhibitor, Murine (40U/μl - NEB, #M0314). The RB-primer-enzyme mix is used to rehydrate two pellets and finally 5μl of MgOAc are added. The complete mix is aliquoted (14.4μl) on top of 5,6 μl of each sample. The RT-RPA protocol was optimised throughout the study. To avoid any confusion, we provide an additional file (Supplementary File 4) with detailed protocols for each experiment presented in this work. All RPA primers used in this study are listed in Supplementary File 3 and were designed following the provided guidelines53.
Cas13 cleavage reaction for lateral flow readout
The reaction mix for Cas13 activity was prepared by combining 4.3 μl of nuclease-free water, 1 μl of cleavage buffer (400mM Tris pH 7.4), 1 μl of LwaCas13a protein diluted in Storage Buffer (SB)53 to a concentration of 126.6 μg/ml, 0.5 μl of crRNA (40 ng/μl), 0.5 μl of lateral flow reporter (IDT, diluted in water to 20 μM), 0.5 μl of SUPERase-In RNase inhibitor (ThermoFisher Scientific, #AM2694), 0.4 μl of rNTP solution mix (25mM each, NEB, #N0466), 0.3 μl of NxGen T7 RNA Polymerase (Lucigen, #30223-2) and 0.5 μl of MgCl2 (120mM). 1 μl of the RT-RPA-amplified product was then added to the mix and, after vortexing and spinning down, the mixture was incubated for 10 minutes at 37°C in a heat block. The Cas13 protocol was optimised throughout the study. To avoid any confusion, we provide an additional file (Supplementary File 4) with detailed protocols for each experiment presented in this work.
Lateral flow readout
Lateral flow detection was performed using commercially available detection strips (Milenia HybriDetect 1, TwistDx, Gieß;en, #MILENIA01). The 10μl-LwaCas13a reactions were transferred to a tube already containing 80 μl of HybriDetect Assay buffer. After vortexing and spinning down the reaction mix, a lateral flow dipstick was added to the reaction tube. The result was clearly readable after one minute. Once the whole reaction volume was absorbed, the dipstick was removed and photographed with a smartphone camera for band intensity quantification performed with the freely available ImageJ image processing program77. The results are shown as intensity ratio (test band/control band) and test were considered positive for value of intensity ratio above 0.2 based on the results shown in Figure S3.
Cas13 cleavage reaction for fluorescence readout
The reaction mix for Cas13 activity was prepared by combining 8.6 μl of nuclease-free water, 2 μl of cleavage buffer (400mM Tris pH 7.4), 2 μl of LwaCas13a protein diluted in Storage Buffer (SB) to a concentration of 126.6 μg/ml, 1 μl of crRNA (40ng/μl), 1 μl of fluorescent reporter (IDT, diluted in water to a final concentration of 4 μM), 1 μl of RNase inhibitor, Murine (NEB, #M0314), 0.8 μl of rNTP solution mix (25mM each, NEB, #N0466), 0.6 μl of NxGen T7 RNA Polymerase (Lucigen, #30223-2) and 1 μl of MgCl2 (120mM). 2 μl of the RT-RPA-amplified product was then added to the mix. The 20μl-LwaCas13a reactions were transferred in 5μl-replicates (4 wells each sample) to a 384-well, round, black-well, clear-bottom plate (Corning, #3544). The plate was briefly spun down at 500g for 15 sec to remove potential bubbles and placed into a pre-heated GloMax® Explorer plate reader (Promega) at 37°C.
Fluorescence readout
Fluorescence was measured every 5 min for 3 h. Data analysis, if not otherwise stated, was performed at the 30-min time-point.
RNAse activity detection assay
In order to check for RNase activity in clinical samples, 10 μl of a negative swab and gargle water sample (Figure 2) were mixed with 10 μl of QuickExtract DNA Extraction Solution with or without RNase Inhibitor, Murine (NEB, M0314) at a final concentration of 4U/μl. The samples were then incubated at 95°C for 5 min. After incubation, RNaseAlert substrate v2 (RNaseAlert Lab Test Kit v2, #4479768, Thermo Fisher Scientific) was added at a final concentration of 200nM. The samples were mixed by vortexing, spun down and incubated at RT for 30 min in the dark. After incubation, the samples were transferred to a 384-well, round, black-well, clear-bottom plate (Corning, #3544) in 5μl-replicates (4 wells each sample). The plate was briefly spun down at 500g for 15 sec to remove potential bubbles and placed into a GloMax Explorer plate reader (Promega). RNaseAlert substrate fluorescence was measured every 5 min for 30 min. Data analysis, if not differently stated, was performed at the 5-min time-point.
Data Availability
All the data will be available after the publication of this article.
AUTHOR CONTRIBUTIONS
BC, RP, and FNP designed the experiments. JPV and AH produced Cas13 protein. BC and RP performed all the experiments using SHERLOCK/ADESSO. PB and BR performed confirmative RT-qPCR on clinical samples using CDC protocol. MK collected the specimens and AGK performed the RT-qPCR on clinical samples using Tib Molbiol, under the supervision of SW. SA quantified the bands of the lateral flow strips. BC and RP analyzed the data and wrote the manuscript. RP, TM and FNP conceived the study and supervised the research. All authors have read and approved the manuscript.
COMPETING INTERESTS
The DKFZ has filed patent applications regarding this diagnostic methodology (EP 20 173 912.5). RP, FNP and BC are inventors on the above-mentioned patent applications.
SUPPLEMENTARY FIGURE LEGENDS

A. LwaCas13a protein purification. The LwaCas13 fusion construct also encodes multiple affinity tags and a protease recognition site at the N terminus of the polypeptide. We have utilized the 6xHIS tag as the basis for our relatively inexpensive purification, while others have developed an alternative protocol based on the Strep-tags53. After expression in Rosetta cells (inducible via the Lac operon), the cells are lysed by sonication and the nucleic acid contained within the lysate is digested. The fusion protein is then purified by nickel-affinity chromatography. The purified fusion protein is digested with SUMO protease, which cleaves the tags and majority of the SUMO site off of the mature protein. The SUMO protease and in-tact affinity tags are then removed from the sample by re-applying the sample to the nickel column, leaving >98% pure Cas13. We also employ a size exclusion chromatography step to remove any aggregated Cas13 protein (not pictured). B. Serially diluted amounts of pure Cas13 were analyzed by coomassie staining after conventional SDS-PAGE, revealing a prominent band at the appropriate molecular weight and only minor contaminants. A serial dilution of BSA was also run as an estimate of protein concentration by densitometry (which was also validated by BCA assay). C. Sensitivity of home-made Cas13 on serial dilutions of an in-vitro-transcribed (IVT) fragment of SARS-CoV-2 S gene in the absence of pre-amplification step. Comparison between fully purified fresh Cas13, partially purified fresh SUMO-Cas13 and fully purified Cas13 stored overnight (o.n.) at 4°C. Both the band intensity ratios (top) and the corresponding lateral flow strips (bottom) are shown. D. Comparison of SHERLOCK sensitivity on the same IVT fragment as in panel C when using either ProtoScript II Retro-Transcriptase (as previously published54) or M-MuLV Retro-Transcriptase in the RT-RPA step.

A. Determination of SHERLOCK sensitivity on serial dilution of SARS-CoV-2 synthetic genome upon optimisation of RT units and RNA input in the RT-RPA reaction with lateral flow readout. B. Comparison of SHERLOCK performance on different genes in SARS-CoV-2 genome by using alternative sets of primers-crRNA targeting N2 (version 1 and version 2), Orf1a61 and S genes on dilutions of a COVID-19 positive sample (sample #6 in Figure 1C). C. Determination of SHERLOCK sensitivity on serial dilutions of SARS-CoV-2 synthetic genome by using the two most sensitive sets of primers-crRNA selected in panel B targeting Orf1a61 and S genes. For panels A, B, C, T = test band; C = control band. For panels A, C, NTC = non template control.
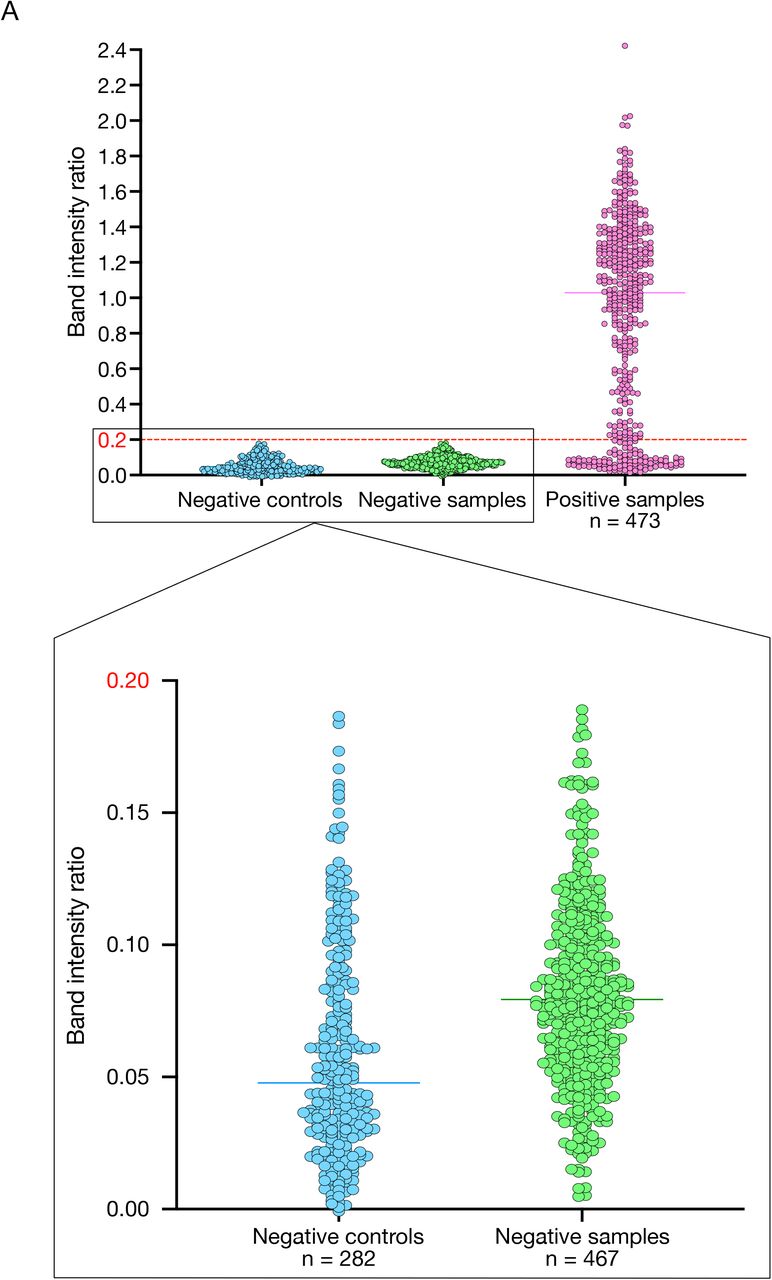
A. The bar plot shows the band intensity ratios of all the negative controls utilised in this study (282, in blue) together with the negative (67(Fig. 2) + 100*4(Fig. 4) = 467, in green) and positive (93(Fig. 2) + 95*4(Fig. 4) = 473, in pink) clinical samples analysed in Figures 2 and 4. These data were used to define a threshold band intensity ratio of 0.2 to distinguish between positive and negative samples.

A. Graphic of SHERLOCK experimental workflow to detect SARS-CoV-2 in unextracted clinical samples with both lateral flow and fluorescence readout. B. Optimisation of SHERLOCK sensitivity with lateral flow readout by increasing the RPA reagents to detect a false negative sample (#L151, Supplementary File 1). The band intensity ratios of the lateral flow strips shown here are plotted in Figure 3C. 1xRPA corresponds to the standard amount of RPA described in the original SHERLOCK protocol53 and 5xRPA corresponds to the optimal amount recommended by the manufacturer. C. Confirmation of the improved SHERLOCK sensitivity with 2xRPA compared to 1xRPA on clinical samples with Ct values close to the LoD from Figure 2 (Supplementary File 1). The band intensity ratios of the lateral flow strips shown here are plotted in Figure 3D. D. Scheme of the experiment and complete measurement of the fluorescence whose results are shown in Figure 3E. E. Time-point analysis of the Cas13 reaction to determine the shortest incubation time required to detect a positive signal with lateral flow readout. The band intensity ratios of the lateral flow strips shown here are plotted in Figure 3F.

A. RPA primers optimisation to amplify the region of SARS-CoV-2 S gene surrounding the B.1.1.7 variant-specific deletion causing ΔHV69-70. Two combinations of the same forward primer with two alternative reverse primers were tested (set 1 and set 2) on serial dilutions of SARS-CoV-2 synthetic genome (Wuhan sequence). Cas13 detection was performed using crRNA HV69-70. Band intensity ratios are shown on the right side. T = test band; C = control band. B. Band intensity ratios of 13 clinical samples carrying either the UK (B.1.1.7) or SA (B.1.351) SARS-CoV-2 variant tested by ADESSO-UK. The corresponding lateral flow strips are shown in Figure 5D and E. The bar plot on the left illustrates the results of the SARS-CoV-2 B.1.1.7 variant detection by ADESSO-UK with crRNA ΔHV69-70. The bar plot on the right illustrates the results of the confirmation of the presence of SARS-CoV-2 B.1.351 variant by ADESSO-UK with crRNA HV69-70. NTC = non template control. C. Schematic of SARS-CoV-2 S gene with annotation of the mutations identified in three patients (clinical samples #11, #12 and #13) carrying the SA variant. The regions of the S gene targeted by ADESSO and ADESSO-UK are indicated in purple and orange, respectively. The presence of the mutation R246I in sample #11, here highlighted in red, disrupts the binding of the RPA forward primer used in ADESSO, thus impeding the amplification of this region and leading to a false negative result.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A and B. Frequency distribution and cumulative frequency distribution, respectively, of the Ct values of all the positive swab samples analysed in this work (n = 211, Supplementary File 1). For both distributions, the bin width is equal to 2 and the R-squared (R2) was calculated for a gaussian distribution.
SUPPLEMENTARY MATERIALS
Positive and negative predictive values, sensitivity and specificity of SHERLOCK on swab samples with (top) and without (bottom) RNA extraction.
ACKNOWLEDGEMENTS
We thank the transport service staff, German Cancer Research Center (DKFZ), for providing excellent transportation services of all the clinical samples from the Medical University Mannheim to the DKFZ. The work was supported by a grant from the Ministry of Science, Research and the Arts of Baden-Württemberg for COVID-19 research (grant agreement no. Kap. 1499 TG 93 to Dr. Riccardo Pecori, Prof. Dr. Nina Papavasiliou (DKFZ) and Prof. Dr. Thomas Miethke (Medical Faculty of Mannheim)). Several schematics presented here were created with Biorender.com.
Footnotes
IMPORTANT NOTE: This protocol should not be used for clinical purposes. Although we have validated the protocol on patient samples, this test is not officially authorized. We hope this protocol will provide some reference points for researchers interested in further advancing Crispr-DX diagnostic platforms. We also welcome researchers to contact us for any assistance.
REFERENCES




